Historien om HS
HS er opkaldt efter den amerikanske læge George Huntington, der beskrev sygdommen i 1872. Hans beskrivelse var baseret på observationer af HS-familier i landsbyen East Hampton på Long Island, New York (USA), hvor Dr. Huntington boede og arbejdede. HS var tidligere kendt som Huntingtons Chorea og Saint Vitus’ dans.
Symptomer & Sygdomsudvikling
HS er en sjælden sygdom, der i Europa findes hos 5 til 10 mennesker ud af 100.000. En tilsvarende hyppighed findes i lande, hvor befolkningen primært er af europæisk oprindelse som for eksempel i USA. HS er mindre hyppig i Asien og Afrika, hvor den er estimeret til at være 1 ud af 100.000 mennesker. Der er samme sandsynlighed for at mænd og kvinder arver forlængelsen i HS-genet og derved udvikler sygdommen. HS er karakteriseret ved en kombination af motoriske (bevægelser), adfærdsmæssige (for eksempel påvirket humør) og kognitive (for eksempel evnen til at forstå) ændringer, men andre symptomer rapporteres også af og til. Debutalder og sværhedsgrad af HS-symptomerne kan variere fra person til person – selv inden for den samme familie – ligesom den hastighed hvormed sygdommen udvikler sig kan variere. Én person har måske tydelige ufrivillige bevægelser, men kun ganske let påvirket adfærd og kognitive evner, mens en anden kan have depression og angst i årevis, inden vedkommende får ufrivillige bevægelser. Mange vil sige, at symptomerne på HS kommer ’snigende’, da det oftest er svært at identificere en specifik dato, hvor symptomerne på HS startede. De fleste, der bærer HS-forlængelsen, udvikler symptomer, når de er imellem 35 og 55 år gamle. Ca. 10% får symptomer inden 20-årsalderen (de har juvenil HS) og tilsvarende får ca. 10% symptomer efter 55-årsalderen. Generelt udvikler HS sig meget gradvist, hvorfor der kan gå mange år før diagnosen bliver stillet. I gennemsnit varer sygdommen 15-20 år fra symptomdebut, men dette varierer fra person til person og afhænger også af kvaliteten af den pleje patienten får. Der er en kompleks sammenhæng imellem debutalderen (den alder, hvor symptomerne starter) ved Huntingtons Sygdom og en række faktorer, der bliver undersøgt i øjeblikket. Når man videnskabeligt betragter store grupper af HS-patienter, kan man se en sammenhæng imellem længden af trinukleotidforlængelsen i HTT-genet og debutalderen (se figur nedenfor). Dette betyder i generelle termer, at jo flere CAG-gentagelser man har, jo tidligere er symptomdebuten (figur nedenfor). For et givet CAG-antal kan variationen i debutalder dog være op til 30 år som vist i figuren nedenfor. Dette skyldes sandsynligvis både variationer i andre gener end HTT-genet (såkaldte genetiske modificerende faktorer) og miljømæssige faktorer som livsstil og kost. Alt i alt betyder det, at det er meget svært at forudsige den præcise debutalder hos en bestemt person, der bærer HS-forlængelsen. Ifølge en klassifikation udviklet af neurolog og HS-specialist Ira Shoulson fra Geargetown University i USA, kan HS inddeles i fem stadier: Når HS starter tidligt i livet (inden man er fyldt 20 år), er de ufrivillige bevægelser (chorea) mindre udtalte i forhold til langsomme bevægelser (bradykinesi) og stivhed i kroppen (dystoni). Tidlige træk ved juvenile HS omfatter markante adfærdsændringer, vanskeligheder med indlæring og tale og faldende præstationer i skolen. Epileptiske anfald rapporteres lejlighedsvis som værende mere almindelige hos unge patienter. Generelt udvikler den juvenile form af sygdommen sig hurtigere, end når den ses hos voksne. Når HS begynder sent i livet, er chorea ofte et mere fremtrædende symptom end langsomhed (bradykinesi) og stivhed (dystoni). Det er sandsynligvis sværere at etablere en familiehistorie, da vedkommendes forældre måske allerede er døde, måske inden de selv viste symptomer på sygdom. Mennesker med HS dør ikke som et direkte resultat af sygdommen, men snarere af medicinske årsager, der opstår som følge af kroppens svækkede tilstand. Disse omfatter lungebetændelse (som tegner sig for en tredjedel af alle dødsfald hos HS-patienter), kvælning, hjertesvigt, hovedskade som følge af fald og ernæringsmæssige mangler. Selvmordsrisikoen er markant forøget og udgør op til 7% af alle dødsfald hos HS-patienter.Hvor almindelig er HS?
Hvad er symptomerne på HS?
De motoriske symptomer, der ses ved HS, er en kombination af chorea, bradykinesi og dystoni, der tydeligt påvirker kropsholdning, balance og gang. Chorea stammer fra det græske ord choreia, der betyder dans, og refererer til de ufrivillige bevægelser, der ofte ses ved HS. At have problemer med at igangsætte frivillige bevægelser (bradykinesi) samt at have vedvarende muskelsammentrækninger, der forårsager en unormal kropsholdning (dystoni), er også almindelige motorsymptomer ved HS. Oculomotoriske (øjenbevægelser) abnormiteter og problemer med at synke er også almindelige og talen bliver mere utydelig og svær at forstå over tid.
De mest almindelige psykiatriske symptomer ved HS er apati, angst, depression, irritabilitet, vredesudbrud, impulsivitet, obsessive kompulsive forstyrrelser, søvnforstyrrelser og social tilbagetrukkethed. Sjældnere er mani og schizofreni inklusiv vrangforestillinger og hallucinationer (se, høre eller føle ting, der ikke eksisterer i virkeligheden) blevet rapporteret. Nogen mennesker med HS kan have selvmordstanker – specielt i de tidlige stadier af sygdommen. De fleste HS-patienter og deres pårørende synes, at de adfærdsmæssige symptomer ved HS er mere problematiske end de motoriske og kognitive symptomer.
HS er karakteriseret ved den gradvise svækkelse af evnen til at forstå, ræsonnere, bedømme og huske. Kognitive symptomer inkluderer langsommere tankegang og problemer med at koncentrere sig, organisere og planlægge ting, tage beslutninger og besvare spørgsmål, ligesom korttidshukkommelsen og evnen til at løse problemer og forstå nye informationer er forringet.
Der er utallige andre ændringer, der kan forekomme under HS sygdomsforløbet som for eksempel mistet appetit, vægttab, tab af selvtillid, tab af sexlyst og inkontinens.Hvornår begynder symptomerne på HS?
Hvad er bestemmende for, hvornår symptomerne starter?
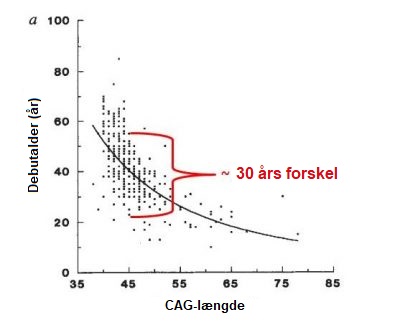
Nature Genetics 4, 398–403 (1993). Hvilke stadier opdeles HS sygdomsforløbet i?
Afviger symptomerne på juvenil HS fra de symptomer,
der ses hos voksne?
Hvad er symptomerne på HS, når sygdommen starter sent i livet?
Dødsårsager
Diagnose & Behandling
HS diagnosticeres ved en kombination af kliniske undersøgelser og en genetisk test. En klinisk diagnose baseres på en persons syge- og familiehistorie samt på standardiserede undersøgelser ved hjælp af kliniske vurderingsværktøjer til at vurdere hyppigheden og sværhedsgraden af HS-symptomerne. Resultatet af den kliniske diagnose bekræftes som regel ved genetisk undersøgelse af HTT -forlængelsen(kaldet en diagnostisk eller bekræftende genetisk test). Hvis en person ikke har symptomer på HS, men er i risiko for at have arvet HTT-forlængelsen, kan vedkommende få undersøgt, om han/hun bærer forlængelsen eller ej uden at have symptomer på sygdom (kaldet en prædiktiv genetisk test).Hvordan diagnosticeres HS?
De kliniske vurderingsværktøjer, der bruges til at diagnosticere HS og kvantificere forskellige aspekter af sygdommen, er ikke ens i alle klinikker i alle lande. Det mest anvendte værktøj er imidlertid “Unified Huntington’s Disease Rating Scale (UHDRS)”, der er opdelt i motoriske, adfærdsmæssige, kognitive og funktionelle underafsnit. Hertil kommer at “Problem Behaviours Assessment for Huntington’s Disease (PBA)” ofte bruges til at vurdere sværhedsgraden og hyppigheden af de adfærdsmæssige ændringer (såsom nedsat stemningsleje, apati og irritabilitet), mens en række forskellige tests som “Mini-Mental State Examination (MMSE)” og “Mattis Dementia Rating Scale” bruges til at supplere den del af UHDRS, der vurderer de kognitive symptomer.Hvilke kliniske vurderingsværktøjer benyttes til at diagnosticere HS?
At leve med en viden om, at man er i risiko for at udvikle HS, er meget bekymrende. Du føler måske, at du ville foretrække at vide med sikkerhed, om du bærer den sygdomsfremkaldende forlængelse i HTT-genet. Hvis det er tilfældet, anbefales det, at du modtager genetisk rådgivning og psykologisk hjælp, fordi du derved får indblik i hvilke muligheder, du har, og du kan diskutere dine bekymringer. Generelt anbefales genetiske undersøgelser ikke til personer under 18 år, da håbet er, at man derved er moden nok til at håndtere bevidstheden om at bære HTT-forlængelsen. I særlige tilfælde kan det imidlertid anses for rimeligt at udføre en bekræftende genetisk test hos børn – hvis de for eksempel viser tegn på juvenil HS – eller hos kvinder, der er under 18 år, hvis de er gravide. Hvis du beslutter dig for at blive testet, vil der blive taget en blodprøve fra en vene i din arm, og dit DNA vil blive ekstraheret i laboratoriet. Lidt afhængig af den enkelte klinik, vil resultatetl være klar to til otte uger senere. Retningslinjerne for hvordan den prædiktive genetiske testprocedure bør forløbe blev opdateret af EHDN Genetic Testing and Counseling Working Group i 2012.Hvad er proceduren ved prædiktiv genetisk testning?
Den genetiske test bestemmer antallet af CAG-gentagelser i HTT -genet. Testen kan afsløre, om du bærer HS-forlængelsen, men den kan ikke fastlægge, hvornår symptomerne på sygdom vil begynde, hvor hurtigt sygdommen vil udvikle sig, eller hvilke symptomer du vil få. Den genetiske test for HS anses for at være så godt som 100% nøjagtig. Resultaterne fra DNA-analysen dobbeltjekkes oftest ved at undersøge to separate blodprøver. Der ud over kan blod fra en forælder til den berørte person (eller, hvis det ikke er tilgængeligt, fra et andet familiemedlem) også undersøges for at bekræfte den oprindelige diagnose.Hvad er det, den genetiske test undersøger?
Ja. Dette gøres ved at benytte en moderne diagnostisk procedure kaldet præimplantations genetisk diagnostik (PGD) – også kendt som fosterscreening – i kombination med in vitro fertilisation (IVF) og involverer screening af fostre, inden de opsættes i livmoderen. Teknikken sikrer, at der kun opsættes fostre, der har arvet to normale kopier af HTT -gen, –selvom en af forældrene bærer HS-mutationen – uanset om det er manden eller kvinden, der er bærer. I nogen lande er PGD dog ikke tilladt på grund af fosterbeskyttelseslovgivning, og det er også vigtigt at vide, at chancen for at fuldføre en graviditet efter PGD/IVF er lavere, end hvis man bliver gravid under “almindelige” omstændigheder. I nogen lande er det muligt at teste ufødte fostre, der er undfanget ad naturlig vej, og dernæst vælge at abortere fosteret, når dets genetiske status er kendt.Er det muligt for en person, der bærer HS-forlængelsen at sikre sig, at vedkommende ikke videregiver HS til deres børn?
Prænatal (før fødslen) diagnose er kun mulig, når de, der beder om at få det foretaget, kan vise, at de opfylder visse medicinske og juridiske kriterier. Disse kriterier kan variere fra land til land. Der er to standardprocedurer for prænatal diagnose. Den første er fostervandsprøven, hvor fostervand, der indeholder celler fra fosteret, opsamles efter fjortende graviditetsuge via en nål igennem moderens mave. Den anden er moderkagebiopsien, der kan udføres tidligere – imellem tiende og trettende graviditetsuge. Denne procedure medfører dog en større risiko for fosteret.Må jeg teste mit ufødte barn?
Der er på nuværende tidspunkt ingen behandlinger, der effektivt kan behandle den underliggende årsag til HS. Dog har basalforskningen og den kliniske forskning øget vores viden om HS i løbet af de seneste år, og der er nu mange studier på vej, der sigter mod at udsætte sygdomsdebut eller nedsætte den hastighed, hvormed sygdommen udvikler sig. Der findes allerede behandlinger, der kan afhjælpe specifikke sygdomssymptomer (symptomatisk behandling) og derved forbedre livskvaliteten for patienterne. Disse behandlinger kan opdeles i farmakologiske (lægemidler) og ikke-farmakologiske behandlinger.Er der nogen behandlinger for HS?
Chorea, badykinesi, irritabilitet, apati, depression, angst og søvnforstyrrelser har alle været rapporteret som de mest frustrerende symptomer ved HS. Der er flere forskellige muligheder for at håndtere disse symptomer med lægemidler. Mange lægemidler kan dog medføre bivirkninger, og nogen modvirker den terapeutiske effekt af andre lægemidler. Desuden kan den samme medicin have forskellig virkning i forskellige personer. Derfor bør behandlinger tilpasses den enkelte patients symptomer og reaktioner på medicinen af en HS-specialist.Hvilke farmakologiske muligheder er der for at behandle
HS-symptomer?
Ikke-farmakologiske behandlinger (som f.eks. psyko-, kognitiv-, tale-, respiratorisk-, fysio- og ergoterapi) kan forbedre de psykiske og fysiske symptomer ved HS. For eksempel er der blevet rapporteret forbedringer i humør, motorisk kontrol, tale, balance, evne til at synke og gå efter disse behandlinger. Det er velkendt, at motion øger både den fysiske og mentale sundhed, forbedrer trivslen generelt, og motion har vist sig også at afhjælpe symptomerne på depression. Der er mere og mere, der tyder på, at motion også bidrager til at bremse udviklingen af de motoriske symptomer ved HS. For eksempel har nogle fysioterapiforløb vist sig at forbedre de motoriske symptomer, gang og balance. EHDNs fysioterapiarbejdsgruppe har udgivet et vejledningsdokument til fysioterapeuter, der arbejder med HS-patienter.Hvordan kan ikke-farmakologiske behandlinger hjælpe?
Fordelene ved en kost fuld af vitaminer, co-enzymer og andre komponenter har været genstand for meget diskussion, men er klinisk aldrig blevet bevist. Men da vægttab er et problem for nogen HS-patienter – især i de sene sygdomsstadier – er det vigtigt at sørge for en sund kost igennem hele sygdomsforløbet. I de sene sygdomsstadier kan det være nødvendigt med en højkaloriediæt. Det kan være nyttigt at blive henvist til en klinisk diætist.Kan en speciel diæt afhjælpe HS-symptomerne?
Arvelighed & Hvad forårsager HS?
HS forårsages af en ændring (en forlængelse) i genet (HTT), der koder for proteinet kaldet huntingtin. Som et resultat af denne forlængelse, translateres (oversættes) genet til en ændret form af proteinet, der forårsager dysfunktion og senere død af nervecellerne (neuronerne) i specifikke områder af hjernen. De specifikke sygdomsmekanismer er meget komplekse, hvilket er i overensstemmelse med de mange forskellige funktioner af huntingtinproteinet. Forskere arbejder på at få en bedre forståelse af de underliggende sygdomsmekanismer for at udvikle sygdomsmodificerende terapier.Hvad forårsager HS?
I 1993 identificerede forskere den mutation, der forårsager HS. HTT -genet findes på kromosom 4 og koder for et protein kaldet huntingtin. Genet indeholder en sekvens af tre nukleotider (de grundlæggende enheder DNA er lavet af), cytosin-adenin-guanin (CAG), der gentages adskillige gange. Denne såkaldte trinukleotid-gentagelse kan variere i længde. Hvis en person har 40 CAG gentagelser eller mere i den ene kopi af HTT -genett, vil vedkommende udvikle HS indenfor en almindelig livslængde – oftest midt i voksenalderen. Da den forlængelse, der forårsager HS, er til stede i alle kroppens celler fra undfangelsen og kan videregives til de efterfølgende generationer, er HS en arvelig sygdom.Hvad er den underliggende årsag til HS?
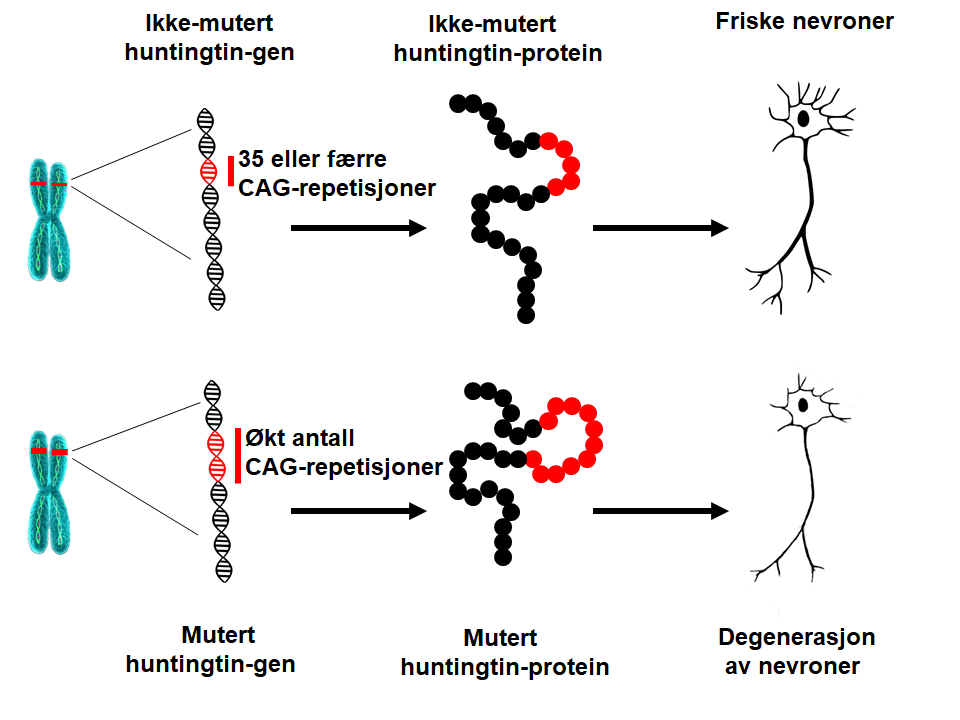
Når antallet af CAG-gentagelser øges, bliver denne del af DNAet mere ustabilt. Dette betyder, at antallet af gentagelser kan øges eller formindskes, når det gives videre til næste generation. Så længe antallet af CAG-gentagelser er under 27 er området stabilt. Hvis antallet af gentagelser er imellem 27 og 35 (de såkaldte intermediære længder), vil personen ikke selv udvikle HS og genet anses for normalt. Dog er CAG-længder på 27 eller flere gentagelser ustabile og kan forlænges, når de gives videre til næste generation, hvilket betyder, at vedkommendes børn har en risiko for at udvikle HS. Personer, der bærer imellem 36 og 39 CAG-gentagelser, udvikler måske HS, men hvis de gør, er det normalt kun meget sent i livet. Derfor kaldes CAG-gentagelser imellem 36 og 39 gråzonen eller længder med nedsat penetrans. Når antallet af CAG-gentagelser er over 39 vil personen udvikle HS inden for almindelig levetid – normalt midt i voksenalderen. I sjældne tilfælde kan CAG-forlængelsen være ekseptionelt lang, hvilket kan føre til sygdomsdebut i teenageårene eller i barndommen (juvenil HS). Patienter, der udvikler symptomer inden 10-årsalderen har ofte mere end 80 CAG-gentagelser.
Hvad betyder længden af CAG-gentagelsen i HTT -genet?
CAG-længde
Forårsager sygdom?
Konsekvenser for efterkommere?
Navn
Under 27
Nej
Ingen
Normal længde
27-35
Nej
Gentagelser med 27 eller flere CAGer kan være ustabile og forlænges, når de gives videre til efterkommere.
Intermediær længde
36-39
Måske
Ja, efterkommere har 50% sandsynlighed for at arve det forlængede gen
Længde med nedsat penetrans
40 og flere
Ja
Ja, efterkommere har 50% sandsynlighed for at arve det forlængede gen
Fuldt penetrant længde
Gener findes på vores kromosomer inde i hver eneste celle i vores krop. Et gen er et stykke DNA, der indeholder koden for et specifikt protein; DNAet transkriberes (kopieres) til messenger RNA (mRNA), der dernæst translateres (oversættes) til protein. Oftest arver alle mennesker to kopier af hvert eneste gen – en kopi fra moderen og en fra faderen. I HS er det vigtige gen HTT -genet, der koder for huntingtinproteinet. Hvis et barn arver en forlænget kopi af HTT -genet, så vil det barn på et tidspunkt også udvikle HS – uanset om forælderen selv har symptomer på sygdommen eller først udvikler det senere.Hvad er et gen?
Proteiner er store molekyler, der består af byggesten kaldet aminosyrer. Den præcise sekvens af aminosyrer i et specifikt protein bestemmes af DNA-sekvensen af det tilsvarende gen. Generne fungerer derved som byggetegninger for cellerne, der herigennem instrueres i, hvordan de skal bygge huntingtinproteinet. Proteinerne er de molekyler, der laver næsten alt arbejdet inde i cellerne – de udfører et stort antal vigtige processer som for eksempel enzymreaktioner eller udøver strukturel støtte. Hvis et protein ikke fungerer normalt eller ikke findes, kan det påvirke cellen og i sidste ende hele organismen og medføre død.Hvad er et protein?
Huntingtinproteinet er et meget stort protein, der laves eller “udtrykkes” i varierende grad i hver eneste celle i den menneskelige krop; de højeste niveauer findes i hjernen. Huntingtin virker til at være et meget vigtigt protein, fordi musefostre dør, hvis ikke proteinet udtrykkes. I den ene ende af huntingtinproteinet er der et område, hvor aminosyren glutamin er gentaget. Denne gentagelse, kaldet polyglutamingentagelsen, består normalt af op til 35 glutaminenheder. Hos mennesker, der bærer HS-forlængelsen, findes der dog mindst 36 glutaminenheder og det er denne polyglutamingentagelse, der er skyld i, at proteinet ikke fungerer normalt.Huntingtinproteinet
HS er en arvelig,dominant sygdom. Det betyder, at en person, der er født med en kopi af det forlængede HTT-gen, vil udvikle HS, selvom han/hun også bærer en normal kopi af genet. Sandsynligheden for, at en person, der bærer HTT-forlængelsen, giver den videre til sine børn er 50% (under forudsætning af, at vedkommende kun bærer én og ikke to forlængede kopier af genet) – uanset om vedkommende har symptomer på sygdom eller ej. Medicinske teknikker kan sikre, at en person, der bærer det forlængede gen, kun videregiver det normale HTT-gen til sine børn. På den anden side vil en person, der ikke har arvet det forlængede HTT-gen, ikke udvikle Huntingtons Sygdom, og vedkommendes børn vil heller ikke være i risiko for at udvikle sygdommen. HS-forlængelsen kan ikke springe en generation over. Det kan dog ske, at en person, der bærer det forlængede HTT-gen, dør før symptomerne opstår, og at vedkommendes børn derfor ikke er klar over, at de er i risiko for at udvikle sygdommen.Hvordan gives HS videre?
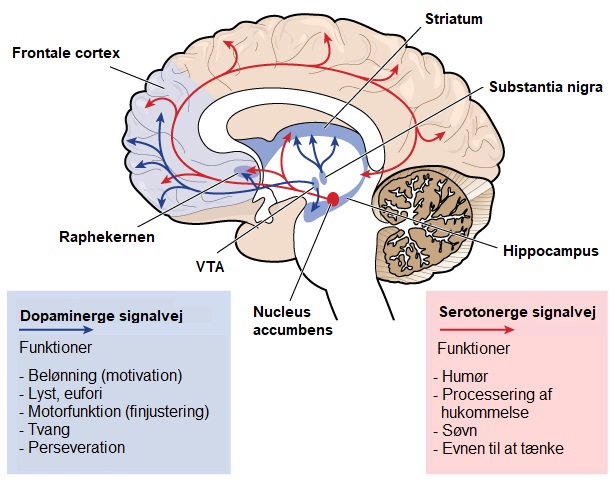
Visse funktioner i hjernen, som evnen til at bevæge sig, tænke og snakke, forringes gradvist i HS, i takt med at vigtige nerveceller bliver beskadigede og dør. Den del af hjernen, der bliver hårdest ramt ved HS, er striatum, som er en del af de basale ganglier og ligger dybt i hjernens centrale region. Striatum er primært involveret i planlægning og styring af bevægelser, men også i mange andre processer, herunder følelserneevnen og i evnen til at tænke. I takt med at HS udvikler sig, påvirker det cortex (den yderste, rynkede del af hjernen), hvilket bidrager til de kognitive symptomer. Over tid forårsager HS generelt atrofi (svind af væv) af hele hjernen, hvilket påvirker det enkelte individs generelle funktionsniveau.Hjerneområder involveret i HS
HS i hverdagen
At blive testet positiv for HS-forlængelsen kan påvirke mange forskellige aspekter af en persons liv, herunder beslutningen om hvorvidt man skal have børn eller ej, hvordan ens fremtid skal planlægges, revurdering af livsprioriteter, finde passende bolig og at informere andre familiemedlemmer om, at de også kan være i risiko for at have arvet sygdommen. Særligt unge voksne vil stå med overvejelser om, hvad et positivt testresultat betyder for deres uddannelse og beskæftigelse. Efterhånden som sygdommen skrider frem, påvirker den gradvist en persons evne til at leve uafhængigt af andre. Arbejde, socialt liv og daglige aktiviteter bliver mere og mere problematiske, og patienter bliver i stigende grad afhængige af hjælp fra slægtninge og sundhedspersonale. Lokale patientforeninger og specialiserede hospitalsafdelinger kan kontaktes, hvis man har brug for hjælp.Hvordan påvirker HS hverdagen?
Effektive strategier, for hvordan man håndterer HS, skal tilpasses det enkelte individ, så man kan tage højde for sygdomsstadie og familiesammenhæng. HS udvikler sig meget langsomt, så der er generelt tid til at tilpasse sig de ændringer, sygdommen medfører. For plejepersonalet og den nærmeste familie kan en bedre forståelse af de adfærdsmæssige og kognitive svækkelser, der er forbundet med sygdommen, måske hjælpe dem til at finde strategier, der imødekommer ændringerne og hjælper dem til at opretholde et godt forhold til personen med HS. Råd og vejledning kan fås ved at kontakte patientforeningen og HS-specialister (sidstnævnte kræver dog en henvisning fra egen læge).Er der strategier for, hvordan man bedre håndterer at leve med HS?
Hvis du kigger på denne side, finder du en liste af “language area coordinators” (landekoordinatorer), der kan hjælpe dig. Alternativt kan du benytte denne kontaktformular.Hvordan kontakter jeg EHDN?
Du skal have en henvisning fra din praktiserende læge til en af de tre specialiserede hospitalsafdelinger i Danmark. Hvis du er i tvivl, kan du kontakte din lokale EHDN local EHDN language coordinator, som vil hjælpe dig.Hvordan får jeg en aftale med en HS-specialist?
Uafhængig hjælp omkring HS kan fås via patientforeningen i dit land.Er der nogen mulighed for at tale med en specialist uden at skulle via hospitalssystemet?
EHDN spiller en nøglerolle i det verdensomspændende kliniske studie Enroll-HD. Det er et observationsstudie, der ikke involverer interventioner. Det betyder, at der ikke som sådan afprøves eksperimentelle behandlinger. Deltagerne i Enroll-HD vurderes klinisk en gang om året, og nogen kan være egnede til at deltage i afprøvning af eksperimentelle behandlinger (der forsøger at påvirke symptomerne ved HS eller at ændre sygdomsforløbet) hvis og når disse bliver tilgængelige. Enroll-HD er tilgængeligt på mange specialiserede HS-afdelinger i hele verden. For at finde ud af, om der er et Enroll-HD center i nærheden af dig, kan du se her eller henvende dig til din EHDN-landekoordinator, som vil kunne informere dig om forskningsaktiviteter i din region. Din lokale patientforening vil kunne give dig generelle oplysninger om at deltage i forskning. For mere information om HS-forskning, se venligst her eller besøg hjemmesiden HDBuzz, der indeholder HS-forskningsnyheder skrevet af HS-forskere i et sprog alle kan forstå og oversat til de fleste sprog.Hvordan bliver jeg involveret i HS-forskning?
Ja, et antal patientforeninger yder støtte til personer og familier, der er berørte af HS. Disse foreninger kan kontaktes direkte. Den Europæiske HS patientforening (The European Huntington’s disease Association, (EHA) fører en liste over patientforeninger i de enkelte lande, der måske kan være nyttig for dig.Er der hjælpegrupper, der er specialiserede i HS?
Kontakt venligst din EHDN landekoordinator eller patientforeningen i dit land, hvis du har andre spørgsmål.