Historia de la EH
La EH debe su nombre a George huntington, un médico americano que describió la enfermedad en 1872. Su descripción se basó en observaciones de familias afectadas por la EH de la aldea de East Hampton, Long Island, Nueva York (EE.UU.), donde vivía y trabajaba el Dr. Huntington. La EH era conocida en el pasado como corea de Huntington y como la danza de San Vito.
Síntomas y progresión
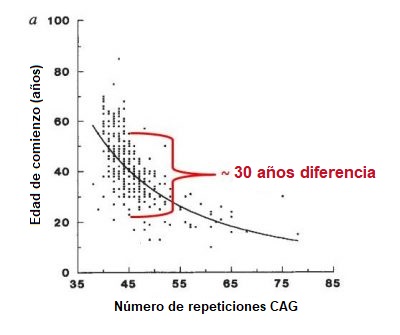
La EH es una enfermedad rara que afecta a entre 5 y 10 personas por cada 100.000 habitantes en la población europea, se encuentra una prevalencia similar en países cuyas poblaciones son principalmente de ascendencia europea, como los EE.UU.. La EH es más rara en países asiáticos y africanos, donde la prevalencia se ha estimado en 1 por cada 100.000 habitantes. Tanto hombres como mujeres tienen la misma probabilidad de heredar la expansión de la EH y desarrollar la enfermedad. La EH se caracteriza por una combinación de alteraciones motoras (movimiento), conductuales (por ejemplo estado de ánimo) y cognitivas (por ejemplo entender) pero también se han descrito otros síntomas. Los síntomas de la EH pueden variar en severidad, edad de comienzo y velocidad de progresión de un individuo a otro – incluso entre los miembros de una misma familia. Una persona puede tener un claro trastorno del movimiento pero síntomas de comportamiento y cognitivos leves, mientras que otro puede tener depresión y ansiedad muchos años antes de que manifieste alteración de movimientos. El inicio de la EH se describe como “insidioso” ya que por lo general es difícil determinar una fecha concreta de comienzo. La mayoría de las personas portadoras de la expansión de la EH desarrollan síntomas en la edad media de la vida, es decir, entre los 35 y los 55 años de edad. Aproximadamente el 10% lo hace antes de los 20 años (tienen EH juvenil) y otro 10% después de los 55 años. En general, la EH se desarrolla muy gradualmente, por lo que puede estar sin diagnosticar durante muchos años. En término medio, la duración de la enfermedad es de 15 a 20 años desde el diagnóstico, pero esto varía de un individuo a otro y también puede depender de la calidad de la atención que recibe el paciente. Los determinantes de la edad de comienzo son complejos y son tema de las investigaciones en curso. Cuando observamos grupos numerosos de pacientes con EH, los científicos encuentran una correlación entre el número de repeticiones de trinucleótidos y la edad de inicio de los síntomas (figura más abajo). En general, cuanto mayor es el número de repeticiones de CAG, más temprano es el inicio de los síntomas (figura más abajo). Sin embargo, si tomamos un número de repetición de CAG, la variabilidad en la edad de inicio puede ser de hasta 30 años, como se muestra en la siguiente figura. Esto se debe probablemente al efecto de otros genes distintos al de la HTT (los denominados modificadores genéticos), y a factores ambientales como el estilo de vida y la dieta. Cuando se tiene en cuenta todo esto es muy difícil predecir con precisión la edad exacta de inicio en cualquier persona que tenga la expansión de la EH. De acuerdo con la clasificación desarrollada por el neurólogo y especialista en la EH, Dr. Ira Shoulson de la Universidad de Georgetown en los EEUU, la progresión de la EH puede dividirse en cinco etapas: Cuando la EH comienza a una edad temprana (antes de los 20 años), los movimientos involuntarios (corea) son menos prominentes que la lentitud del movimiento (bradicinesia) y la rigidez (distonía). Los primeros rasgos de la EH juvenil incluyen marcados cambios de comportamiento, dificultades en el aprendizaje y el habla, y disminución en el rendimiento escolar. En ocasiones aparecen crisis epilépticas, siendo más frecuentes en pacientes jóvenes. En general, la forma juvenil de la enfermedad progresa más rápidamente que la forma adulta. Cuando la EH comienza a una edad tardía, la corea tiende a ser más prominente que la lentitud o la distonía. En estos casos es probable que sea más difícil de encontrar antecedentes familiares porque los padres del paciente habrán fallecido, incluso antes de que ellos mismos mostraran signos de la enfermedad. Las personas con EH no fallecen a consecuencia directa de la enfermedad, sino de problemas médicos que surgen como resultado del debilitamiento corporal. Estos problemas incluyen neumonía (causa de fallecimiento de un tercio de los pacientes con EH), atragantamiento, infarto cardíaco, traumatismo cerebral debido a caídas y deficiencias nutricionales. El riesgo de suicidio está considerablemente elevado, siendo responsable del 7% de los fallecimientos de todos los pacientes.¿Es frecuente la EH?
¿Cuáles son los síntomas de la EH?
Los signos motores de la EH son una mezcla de corea, bradicinesia y distonía, que afectan notablemente la postura, el equilibrio y la marcha. Corea proviene de la palabra griega choreia, que significa danza y se refiere a los movimientos involuntarios a menudo vistos en la EH. Otros signos motores comunes en la EH son tener problemas para iniciar movimientos voluntarios (bradicinesia) y contracciones musculares sostenidas que causan posturas anormales y torsiones o torsiones (distonía). También son comunes las alteraciones oculomotoras (movimientos oculares) y las dificultades de deglución, y el habla se vuelve más difícil de entender con el tiempo.
Los síntomas psiquiátricos más comunes de la EH son la apatía, la ansiedad, la depresión, la irritabilidad, los estallidos de ira, la impulsividad, los comportamientos obsesivo-compulsivos, los trastornos del sueño y el aislamiento social. Más rara vez, se ha reportado manía y esquizofrenia – incluyendo delirios (falsas creencias) y alucinaciones (ver, oír o sentir cosas que no existen). Los individuos afectados pueden experimentar pensamientos suicidas, especialmente en las primeras etapas de la enfermedad. La mayoría de los pacientes con EH y sus cuidadores perciben que las alteraciones del comportamiento causadas por la enfermedad son más angustiosas que los cambios motores o cognitivos.
La EH se caracteriza por el deterioro gradual de la comprensión, el razonamiento, el juicio y la memoria. Los síntomas cognitivos incluyen un pensamiento más lento y dificultad para concentrarse, organizarse, planificar, tomar decisiones y responder preguntas, así como déficit de memoria a corto plazo y resolución de problemas y una capacidad deteriorada para añadir y comprender nueva información.
Hay una serie de cambios que pueden ocurrir durante el curso de la EH, incluyendo pérdida del apetito, pérdida del peso, pérdida de la autoestima, pérdida del deseo sexual e incontinencia urinaria y fecal.¿Cuándo aparecen los síntomas de la EH?
¿Qué determina la edad de comienzo de los síntomas?
¿Cuáles son los diferentes estadíos de la progresión de la EH?
¿Los síntomas de la EH juvenil son diferentes a los del adulto?
¿Cuáles son los síntomas cuando la EH aparece a una edad más tardía?
Causas de fallecimiento
Diagnóstico & Tratamiento
La EHD se diagnostica mediante una combinación de evaluaciones clínicas y una prueba genética. El diagnóstico clínico se basa en la historia médica y familiar de una persona, así como en los exámenes estándar que hacen uso de escalas de evaluación clínica para evaluar la frecuencia y gravedad de los síntomas de la EH. Los resultados del diagnóstico clínico generalmente se confirman mediante el análisis genético para la expansión de HTT (conocido como diagnóstico o análisis genético confirmatorio). Si una persona no presenta ningún síntoma, pero está en riesgo de la enfermedad, puede realizarse un análisis genético asintomático (conocido como análisis genético predictivo) que determinará si es portador o no de la expansión.¿Cómo se diagnostica la EH?
Las herramientas de evaluación clínica utilizadas para diagnosticar la EH y medir aspectos de su presencia no son las mismas en todos los centros de todos los países. Sin embargo, la herramienta más comúnmente utilizada es la Unified Huntington’s Disease Rating Scale (UHDRS), que se divide en subsecciones motoras, conductuales, cognitivas y funcionales. Además, la evaluación de los problemas del comportamiento en la enfermedad de Huntington (PBA) se utiliza a menudo para evaluar la gravedad y la frecuencia de las alteraciones del comportamiento (como el estado de ánimo depresivo, la apatía y la irritabilidad), mientras que una variedad de pruebas como el Mini-Mental State Examination (MMES) y la Escala de Evaluación de la Demencia de Mattis se usan para complementar la subsección de la UHDRS que evalúa las alteraciones cognitivas.¿Qué pruebas clínicas se utilizan para diagnosticar la EH?
Vivir sabiendo que usted está en riesgo de padecer EH puede ser muy preocupante. Usted puede sentir que preferiría saber con certeza si es portador de la expansión. En ese caso, el consejo genético y el apoyo psicológico son muy recomendables, ya que le permiten conocer sus opciones y hablar de sus preocupaciones. En general, la prueba predictiva no es recomendable antes de los 18 años – la edad en la que, se espera, que una persona ya tiene la madurez para hacer frente los resultados de portar la expansión. En casos excepcionales, sin embargo, puede considerarse razonable realizar la prueba genética confirmatoria en niños – si muestran signos de EH juvenil, por ejemplo – o en mujeres menores de 18 años si están embarazadas. Si decide hacerse la prueba, le tomarán una muestra de sangre y extraerán su ADN de ella en el laboratorio. Dependiendo del servicio local, el resultado estará listo entre dos a ocho semanas. Las Normas Eticas para realizar el análisis genético predictivo fueron actualizadas por el Grupo de Trabajo sobre el Consejo y el Análisis Genético en 2012.¿Cuál es el procedimiento para realizar el análisis genético predictivo?
La prueba genética determina el número de repeticiones CAG en el gen HTT. El análisis puede desvelar si usted es portador de la expansión de la EH, pero no puede determinar cuándo comenzará la enfermedad, la velocidad de la progresión o qué síntomas podría desarrollar. La prueba genética para la EH se considera que tiene una exactitud cercana al 100%. Los resultados del análisis de ADN generalmente se verifican utilizando dos muestras de sangre diferentes. Además, también se puede utilizar una muestra de sangre de un padre de la persona afectada (o, si no está disponible , de otro miembro de la familia) para proporcionar la confirmación del diagnóstico original.¿Qué detecta el análisis genético?
Sí. Esto se puede conseguir utilizando un procedimento diagnóstico moderno llamado diagnóstico genético pre-implantacional (DGP) – también conocido como estudio embrionario – que se utiliza en combinación con técnicas de fertilización in vitro(FIV) y que implica el estudio de los embriones antes de ser implantados en el útero. La técnica asegura que sólo los embriones con copias normales del gen serán implantados. Por lo tanto, incluso si uno de los padres es portador de la mutación de la EH, el DGP hace posible que esa pareja conciva un hijo que no sea portador del gen mutado de la EH, – con independencia de que el portador sea el hombre o la mujer. De todos modos el DGP no está permitido en todos los países por las leyes de protección de los embriones y también hay que tener en cuenta que la probabilidad de embarazo tras una técnica DGP/FIV es más baja que con la concepción «natural». En algunos países también es posible hacer un análisis del feto concebido de manera natural y abortar en caso de que el feto sea portador.¿Es posible que una persona portadora de la expansión de la EH no la
pase a su hijo?
El diagnóstico prenatal (antes del nacimiento) sólo está disponible cuando los solicitantes pueden demostrar que su caso cumple ciertos criterios médicos y legales, que son específicos de cada país. Existen dos procedimientos estándar para el diagnóstico prenatal. La primera es la amniocentesis (también llamada prueba del líquido amniótico), en la cual se recoge líquido amniótico que contiene las células fetales con una aguja insertada a través de la pared abdominal de la madre, generalmente después de la 14a semana de embarazo. El segundo procedimiento es la recogida de vellosidad coriónica, que implica la recolección de una muestra de las vellosidades coriónicas (tejido placentario) y se puede realizar antes – entre las semanas 10 y 13 de embarazo. Sin embargo, es más arriesgado para el feto.¿Puedo hacer un análisis del feto?
Actualmente no hay terapias que puedan tratar eficazmente las causas subyacentes de la EH. Sin embargo, la investigación básica y clínica ha aumentado dramáticamente nuestro conocimiento sobre la EH en los últimos años, y muchos studies are now underway that are investigating its pathogenesis, with a view to identifying drugs that will postpone disease onset or slow its progression. Treatments are already available that alleviate certain symptoms of the disease (symptomatic treatments), and so improve patients’ quality of life. These are divided into pharmacological (drug) and non-pharmacological (non-drug) treatments.¿Hay algún tratamiento para la EH?
Corea, bradicinesia, irritabilidad, apatía, depresión, ansiedad y trastornos del sueño son los síntomas más angustiosos de la EH. Hay varias opciones para manejar estos síntomas usando fármacos. Sin embargo, muchos medicamentos pueden causar efectos secundarios, y algunos pueden contrarrestar los efectos terapéuticos de otros. Además, la misma medicación puede tener efectos diferentes en individuos diferentes. El tratamiento debe ser personalizado por un especialista experimentado en EH, de acuerdo con los síntomas del paciente y su respuesta a los fármacos en cuestión.¿Cuáles son los fármacos para tratar los síntomas de la EH?
Los tratamientos no farmacológicos (como la psicoterapia y la rehabilitación cognitiva, el ejercicio físico, la logopedia, la terapia respiratoria y ocupacional) pueden mejorar los síntomas psicológicos y físicos de la EH. Por ejemplo, se han encontrado mejoras en el estado de ánimo, control motor, el habla, el equilibrio, la deglución y la marcha después de estas terapias. Es bien sabido que el ejercicio físico mejora la salud física y mental, mejorando el bienestar general, y el ejercicio se ha demostrado que alivia los síntomas de la depresión. Se van sumando pruebas de que también puede ayudar a retrasar la progresión de las alteraciones motores en la EH. Por ejemplo, algunos programas de fisioterapia han mostrado beneficios en los síntomas motores, la marcha y el equilibrio. Elgrupo de trabajo de Fisioterapia del EHDN ha publicado una guía orientativa para los fisioterapeutas que trabajan con pacientes con EH.¿Los tratamientos no farmacológicos pueden ayudar?
Los beneficios de una dieta rica en vitaminas, co-enzimas y otros compuestos han sido objeto de mucha discusión, pero aún deben ser probados clínicamente. Sin embargo, como la pérdida de peso es un problema para algunos pacientes con EH, especialmente en las últimas etapas de la enfermedad, es importante asegurar una dieta saludable a lo largo de la enfermedad. En etapas posteriores una dieta alta en calorías puede ser necesaria. La remisión a un dietista puede ser útil.¿Una dieta especial puede mejorar los síntomas de la EH?
Herencia & y causa de la EH
La EH está causada por un cambio (expansión) en el gen (HTT) que codifica una proteína llamada huntingtina. Como resultado de esta expansión, el gen se traduce en una forma alterada de la proteína, algo que resulta en el mal funcionamiento y la muerte de las células nerviosas (neuronas) en áreas específicas del cerebro. Los mecanismos exactos de la enfermedad son multifacéticos y altamente complejos, de acuerdo con las múltiples funciones de la proteína huntingtina. Los investigadores están trabajando por conseguir una mejor comprensión de los mecanismos subyacentes que causan la enfermedad para desarrollar tratamientos para la enfermedad.¿Qué causa la EH?
En 1993, los científicos identificaron la mutación que causa la EH. El gen HTT está situado en el cromosoma 4 y codifica una proteína llamada huntingtina. El gen contiene una secuencia de tres nucleótidos (las unidades básicas del ADN), citosina-adenina-guanina (CAG), que se repite varias veces. Esta supuesta repetición de trinucleótidos puede variar en longitud. Si una persona tiene 40 o más repeticiones CAG en una copia del gen HTT, desarrollará EH a lo largo de su vida – normalmente en la edad media de la vida. Puesto que la expansión que causa la EH está presente en todas las células del cuerpo desde la concepción y puede transmitirse a las siguientes generaciones, la EH es una enfermedad hereditaria.¿Cuál es la causa subyacente de la EH?
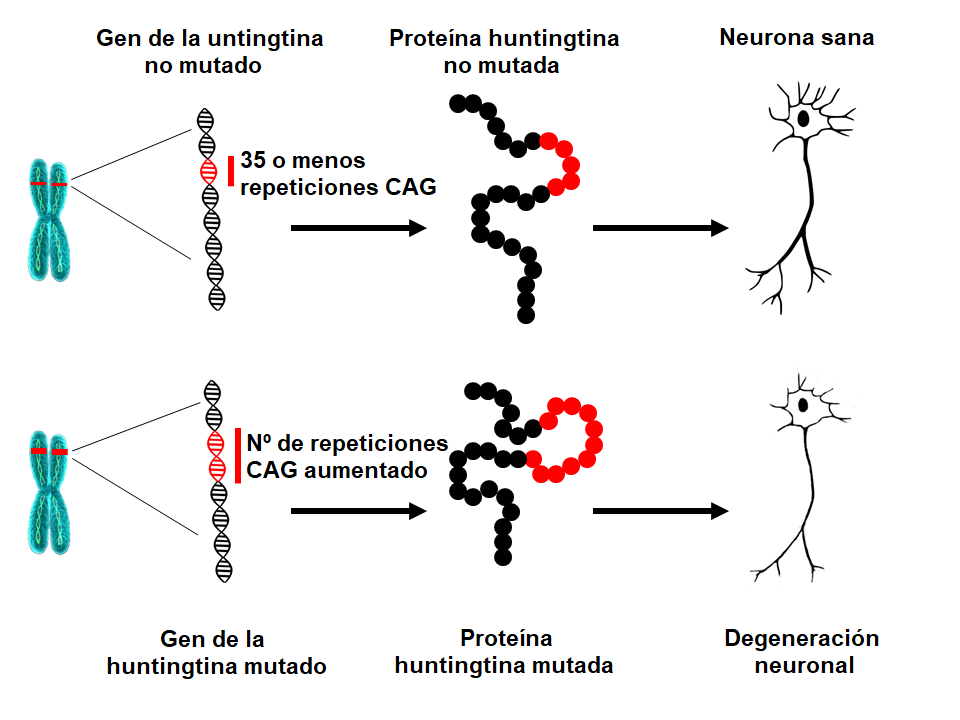
Según aumenta el número de CAG , esa sección particular de ADN se vuelve más inestable. Esto significa que el número de repeticiones en esta sección puede aumentar o disminuir cuando se transmite a la siguiente generación. Cuando el número de repeticiones de CAG en el gen de la HTT es inferior a 27, la sección es estable. Si el número de repeticiones está entre 27 y 35 (llamado rando intermedio de repeticiones), ese individuo no va a desarrollar la EH y el gen se considera normal. Sin embargo, un número de repetición de CAG de 27 o más es inestable y tiende a aumentar cuando pasa a la siguiente generación, lo que significa que los niños tienen riesgo de desarrollar la EH. Los individuos con un número de repeticiones CAG entre 36 y 39 pueden desarrollar EH, pero a una edad más avanzada, si llegan a desarrollarla. Esto se conoce como número de repeticiones de penetrancia reducida. Cuando el número de repeticiones de CAG es superior a 39, una persona desarrollará EH a lo largo de su vida – con frecuencia en la edad media. En raras ocasiones la expansión de CAG puede ser excepcionalmente larga, lo que lleva a un inicio de la enfermedad en la adolescencia o la niñez (EH juvenil). Los pacientes que desarrollan la enfermedad antes de los 10 años a menudo tienen más de 80 repeticiones CAG.
¿Qué significa el número de HTT repeticiones de CAG?
Número de repeticiones CAG ¿Causa enfermedad? ¿Consecuencia para la descendencia? Resultado
Por debajo de 27
No
Ninguna
Número normal de repeticiones
27-35
No
27 o más repeticiones puede ser inestable y dicho número puede aumentar en la siguiente generación
Número de repeticiones intermedio
36-39
Puede ser
Sí, la descendencia tiene un 50% de probabilidades de heredar el gen expandido
Número de repeticiones de penetrancia reducida
40 o más
Sí, la descendencia tiene un 50% de probabilidades de heredar el gen expandido
Sí, la descendencia tiene un 50% de probabilidades de heredar el gen expandido
Número de repeticiones de penetrancia completa
Los genes se encuentran en los cromosomas dentro de cada célula de nuestro cuerpo. Un gen es un segmento de ADN que contiene el código para fabricar una proteína particular; el ADN se transcribe en ARN mensajero (ARNm), que luego se traduce en la proteína. Como norma general, cada uno hereda dos copias de cada gen, una de la madre y una del padre. En la EH el gen afectado es el gen de la HTT, que codifica la proteína huntingtina. Cuando un niño hereda una versión expandida del gen de la HTT entonces ese niño desarrollará la EH. Puede que uno de los padres ya tenga la enfermedad o que la desarrolle más tarde.¿Qué es un gen?
Las proteínas son grandes moléculas compuestas de bloques de construcción llamados aminoácidos. La secuencia exacta de aminoácidos en una proteína dada es determinada por la secuencia de ADN del gen correspondiente. Los genes por lo tanto funcionan como planos – conjunto de instrucciones que le dicen a las células cómo construir las proteínas específicas. El gen de la HTT contiene instrucciones sobre cómo construir la proteína huntingtina. Las proteínas son las moléculas que hacen el trabajo dentro de las células, que realizan un gran número de procesos esenciales, tales como reacciones enzimáticas o soporte estructural. Si una proteína funciona mal o se pierde debido a una expansión en el gen que la codifica, luego puede afectar a la célula y, en última instancia, a todo el organismo, a veces causando una enfermedad.¿Qué es una proteína?
La proteína huntingtina es una proteína muy grande que se produce o «expresa» en diversos grados en cada célula del cuerpo humano; los niveles más altos se encuentran en el cerebro. La huntingtina parece ser una proteína muy importante dado que la ausencia de ella produce la muerte en los embriones de ratón. En un extremo de la proteína huntingtina hay un trozo que contiene repeticiones de un determinado aminoácido llamado glutamina. Esta característica distintiva, conocida como repetición de poliglutaminas, normalmente consta de hasta 35 unidades de glutamina. Las personas portadoras de la expansión de la EH, sin embargo, tienen al menos 36 repeticiones y esta expansión de poliglutaminas da como resultado una proteína que funciona mal.La proteína huntingtina
La EH es una enfermedad hereditaria dominante. Esto significa que una persona nacida con una copia del gen de la HTT mutada desarrollará EH a pesar de que él o ella también lleve una copia normal del gen. Un portador de la expansión de la EH, ya sea sintomático o no, puede pasar a su descendencia una copia normal o una copia mutada del gen con una probabilidad del 50% de cada uno (siempre que él o ella sólo lleve una copia de la expansión del gen de la HTT). Algunas técnicas médicas pueden asegurar que un individuo afectado solo pase el gen de la HTT normal a sus hijos. Por el contrario, una persona que no ha heredado el gen de la HTT mutado no desarrollará la enfermedad, y sus hijos no estarán a riesgo. La expansión de la EH no puede saltarse una generación. Sin embargo, puede suceder que un portador de la expansión muera antes de mostrar síntomas, y que sus hijos no se den cuenta de que están a riesgo de desarrollar la enfermedad.¿Cómo se transmite la EH?
Ciertas funciones del cerebro como la capacidad de moverse, pensar y hablar poco a poco se deterioran en la EH porque las células nerviosas se deterioran y mueren. La parte del cerebro más afectada por la EH es el cuerpo estriado, que es un componente de los ganglios basales y está situado en la región profunda del cerebro. El estriado está implicado principalmente en la planificación y control de movimientos, pero también en muchos otros procesos, incluyendo la cognición y las emociones. A medida que progresa la EH, se afecta la corteza (la parte arrugada más externa del cerebro), contribuyendo al deterioro cognitivo. En general, la EH provoca la atrofia de todo el cerebro con el tiempo, altera la capacidad funcional general del individuo.Areas cerebrales implicadas en la EH
EH en el día a día
Ser portador de la expansión de la EH puede afectar a diferentes aspectos de la vida de una persona, incluyendo la decisión de tener hijos, pensar en el futuro, repensar las prioridades, negociar la vivienda apropiada e informar o no a otros familiares que también pueden estar a riesgo de la enfermedad. Las personas más jóvenes en particular pueden necesitar considerar las implicaciones de un resultado positivo para su educación, formación y empleo. Según avanza la enfermedad, la independencia de una persona se ve poco a poco afectada. El trabajo, la vida social y en general las actividades cotidianas son problemáticas y los pacientes son cada vez más dependientes de la ayuda de familiares y de profesionales sanitarios y de atención social. Puede ponerse en contacto con los grupos locales de apoyo a pacientes y los centros clínicos para solicitar apoyo.¿Cómo afecta la EH a la vida diaria?
Las estrategias eficaces para hacer frente a la EH han de ser personalizadas y dependen de la persona afectada, la etapa de la enfermedad y el contexto familiar. La EH va avanzando muy lentamente, por lo que en general hay tiempo para adaptarse a los cambios que trae consigo. Para los cuidadores y los seres queridos les puede ser útil una mejor comprensión de las alteraciones conductuales y cognitivas asociadas a la enfermedad para desarrollar estrategias para asimilar estos cambios y mantener una buena relación con la persona afectada. Los especialistas en EH y los grupos de apoyo de pacientes les pueden proporcionar información útil y consejos.¿Hay alguna estrategia para afrontar mejor la EH?
Usted puede consultar esta página esta página donde encontrará un listado de coordinadores locales que le ayudarán. También puede utilizar el contacto que encontrará aquí.¿Cómo me puedo poner en contacto con el EHDN?
Puede conseguir una cita a través de su médico de cabecera general, o poniéndose en contacto con el coordinador local del EHDN quien le ayudará.¿Cómo pedir cita con un especialista?
Puede obtener información sobre la EH de los grupos de ayuda de pacientes en su país¿Puedo hablar con un especialista sin tener que ir a una clínica?
El EHDN desempeña un papel clave en el estudio clínico mundial llamado Enroll-HD. Es un estudio observacional que no implica ninguna intervención. Esto significa que no se prueba ningún tratamiento experimental propiamente. Los participantes se someten a evaluaciones clínicas durante sus visitas anuales y pueden ser elegibles para participar en ensayos clínicos de tratamientos sintomáticos o modificadores de la enfermedad cómo y cuándo éstos estén disponibles. Se puede participar en el estudio Enroll-HD a través de muchos centros donde se estudia la EH alrededor del mundo. Para saber si hay algún centro cerca de usted, por favor consulte aquí o contacte con su coordinador local del EHDN, quién podrá informarle sobre las actividades científicas en su región. Los grupos de apoyo local de los pacientes serán capaces de proporcionarle información general sobre la participación en la investigación. Para obtener más información sobre la investigación en la EH, por favor, consulte aquí o visite la página web HDBuzz con novedades sobre la investigación en la EH escrito por investigadores de la EH en idioma sencillo y traducido a varios idiomas.¿Cómo puedo participar en la investigación de la EH?
Sí, hay una serie de grupos de apoyo de los pacientes que proporcionan apoyo a individuos y familias afectadas por la EH. Puede ponerse en contacto con ellos a través de su médico de cabecera o el especialista en EH o directamente. La Asociación Europea de Enfermedad de Huntington (EHA) y la Asociación Internacional de Huntington (IHA) tienen una de los pacientes que puede serle útil.¿Hay grupos de apoyo especializados en la EH?
Para cualquier otra duda, póngase en contacto con su coordinador local del EHDN o su grupo local de apoyo de los pacientes.