Historien om HS
HS er oppkalt etter George Huntington, en amerikansk lege som beskrev sykdommen i 1872. Hans beskrivelse var basert på observasjoner av HS-berørte familier fra landsbyen East Hampton, Long Island, New York, USA, hvor Dr Huntington bodde og jobbet. HS var tidligere kjent som Huntingtons chorea, Saint Vitus’ dans eller Setesdalsrykkja.
Symptomer & sykdomsprogresjon
HS er en sjelden sykdom som i Europa rammer 5-10 personer per 100 000. En lignende utbredelse finnes i land der befolkningen primært er av europeisk avstamning, som for eksempel USA. HS er mindre vanlig i asiatiske og afrikanske land, hvor forekomsten er estimert til 1 person per 100 000. Menn og kvinner har like stor sannsynlighet for å arve forlengelsen av HS-genet og å utvikle sykdommen. HS karakteriseres av en kombinasjon av motoriske (bevegelsesforstyrelser), atferdsmessige (for eksempel humørendringer) og kognitive (for eksempel evnen til å forstå) symptomer, men andre symptomer kan også forekomme. Debutalder, alvorlighetsgrad og hvor raskt HS- symptome utvikler seg kan variere fra person til person – også innen samme familie. En person kan ha markante bevegelsesforstyrrelser, men bare milde atferdssymptomer og kognitiv svekkelse, mens en annen kan lide av depresjon og angst i mange år innen vedkommende får ufrivillige bevegelser. Oppstarten av HS beskrives ofte som “snikende”, siden det vanligvis er vanskelig å bestemme en eksakt dato for når symptomene på HS startet. De fleste personer som bærer HS-forlengelsen, utvikler symptomer i midten av voksenlivet, det vil si mellom 35 og 55 år. Om lag 10% gjør det før de fyller 20 år (de har juvenil HS) og ytterligere 10% etter fylte 55 år. Generelt utvikler HS seg veldig gradvis, slik at sykdommen kan gå udiagnostisert i mange år. I gjennomsnitt varer sykdommen i 15 til 20 år fra symptomdebut, men dette varierer fra person til person og kan også avhenge av kvaliteten på pleien som pasienten får. De bestemmende faktorende som avgjør tidspunktet for sykdomsutbrudd er komplekse og gjenstand for pågående undersøkelser. Når man undersøker store grupper av HS-pasienter, finner forskerne en sammenheng mellom antallet CAG trinukleotid-repetisjoner og debutalder (se figur nedenfor). I generelle termer betyr dette at jo høyere antallet CAG repetisjoner er, jo tidligere starter symptomene (figur nedenfor). Men, for hvert gitt CAG-antall kan variasjonen i debutalder være opptil 30 år, som vist med figuren nedenfor. Dette skyldes trolig effektene av andre gener enn HTT-genet (såkalte genetisk modifiserende faktorer), og miljøfaktorer som livsstil og kosthold. Alt i alt innebærer dette, at det er svært vanskelig å forutsi debutalderen til en person som bærer HS-genforlengelsen. I følge en klassifisering utviklet av nevrolog og HS-spesialist Ira Shoulson fra Georgetown University i USA, kan progresjonen av HS deles inn i fem faser: When HD starts early in life (before the age of 20), involuntary movements (chorea) are less prominent as a symptom than slowness of movement (bradykinesia) and stiffness (dystonia). Early features of juvenile HD include marked behavioural changes, difficulties with learning and speech, and decline in performance at school. Epileptic seizures are occasionally reported, being more common in young patients. In general, the juvenile form of the disease progresses more rapidly than the adult form. Når HS starter sent i livet er chorea ofte et mer fremtredende symptom enn langsomhet (bradykinesi) eller stivhet (dystoni). Det er sannsynligvis vanskeligere å etablere en familiehistorikk, fordi personens foreldre allerede er døde, kanskje før de selv viste tegn på sykdommen. Personer med HS dør ikke som et direkte resultat av sykdommen, men heller fra medisinske problemer som oppstår som følge av kroppens svekkede tilstand. Disse inkluderer lungebetennelse (som står for en tredjedel av alle dødsfall hos HS-pasienter), kvelning, hjertesvikt, hodeskader som følge av fall og ernæringsmessige mangler. Selvmordsrisikoen er markant forhøyet, og står for opptil 7% av alle dødsfall hos HS-pasienter.Hvor vanlig er HS?
Hva er symptomene på HS?
De vanligste psykiatriske symptomene på HS er apati, angst, depresjon, irritabilitet, utbrudd av sinne, impulsivitet, obsessiv-kompulsiv oppførsel (tvangstanker), søvnforstyrrelser og sosial tilbaketrekking. Mer sjelden har mani og schizofreni – inkludert vrangforestillinger og hallusinasjoner (se, høre eller føle ting som egentlig ikke finnes) blitt rapportert. Noen personer med HS kan få selvmordstanker, spesielt i tidlige stadier av sykdommen. De fleste HS-pasienter og deres omsorgspersoner synes at atferdssymptomene er mer plagsomme enn de motoriske og kognitive symptomene.
HS karakteriseres av en gradvis svekkelsen av evnen til å forstå, resonnere, bedømme og huske. Kognitive symptomer inkluderer langsommere tankegang og problemer med konsentrasjon, organisering, planlegging, ta avgjørelser og svare på spørsmål. Samt manglende korttidshukommelse, evne til å løse problemer og en svekket evne til å ta til seg og forstå ny informasjon.
Det er en rekke andre endringer som kan oppstå i løpet av sykdomsforløpet, inkludert tap av matlyst, vekttap, svekket selvtillit, svekket seksuallyst, og inkontinens for urin og avføring.Når starter symptomene på HS?
Hva bestemmer alderen for når symptomene starter?
Hvilke sykdomsstadier finnes ved HS?
Avviker symptomene på juvenil HS fra de sykdommene som ses
hos voksne?
Hva er symptomene på HS, når sykdommen starter sent i livet?
Dødsårsaker
Diagnose & Behandling
HS diagnostiseres ved en kombinasjon av kliniske undersøkelser og en genetisk test. En klinisk diagnose baseres på en persons syke- og familiehistorie, samt på standardiserte undersøkelser ved hjelp av kliniske vurderingsverktøy for å vurdere hyppigheten og alvorlighetsgraden av symptomene på HS. Resultatene av den kliniske diagnosen bekreftes vanligvis ved genetisk screening av HTT-forlengelsen (kjent som diagnostisk eller bekreftende genetisk testing). Hvis en person ikke har symptomer på HS, men er i risiko for å ha arvet sykdommen, kan vedkommende få undersøkt om han/hun bærer forlengelsen eller ikke (kjent som prediktiv genetisk testing).Hvordan diagnostiseres HS?
De kliniske vurderingsverktøyene som brukes til å diagnostisere HS og måle forskjellige aspekter av sykdommen, er ikke de samme på alle sykehus i alle land. Det mest brukte verktøyet er imidlertid “Unified Huntington’s Disease Rating Scale (UHDRS)”, som er delt inn underavsnitt innen motorikk, atferd, kognisjon og funksjon. I tillegg brukes ofte “Problem Behaviours Assessment for Huntington’s Disease (PBA)” for å vurdere alvorlighetsgraden og hyppigheten av de atferdsmessige endringene (som nedsatt stemningsleie, apati og irritabilitet), mens en rekke forskjellige tester, som ”Mini-Mental State Examination (MMSE)” og ”Mattis Dementia Rating Scale” brukes til å supplere den delen av UHDRS som vurderer de kognitive symptomene.Hvilke kliniske vurderingsverktøy benyttes til å diagnostisere HS?
Å leve med en viten om at du er i faresonen for å utvikle HS kan være svært bekymringsfull. Du føler kanskje at du ville foretrekke å vite med sikkerhet, om du er bærer av den sykdomsfremkallende forlengelsen av HTT-genet. Hvis det er tilfelle, anbefales det at du mottar genetisk rådgivning og psykologisk hjelp, slik at du kan utforske dine alternativer og diskutere eventuelle bekymringer. Generelt anbefales ikke prediktiv testing for de som er yngre enn 18 år – alderen da en forhåpentligvis er moden nok til å takle å få vite om man har forlengelsen eller ikke. I unntakstilfeller kan det imidlertid anses å være rimelig å utføre den bekreftende genetiske testen hos barn – hvis de for eksempel viser tegn på juvenil HS – eller hos kvinner under 18 år, hvis de er gravide. Hvis du bestemmer deg for å bli testet, vil en blodprøve bli tatt fra en vene i armen din, og DNA vil bli ekstrahert i laboratoriet. Resultatet er klart etter to til åtte uker, avhengig av den enkelte klinikk som utfører testen. Retningslinjer for den prediktive genetiske testprosedyren ble oppdatert av EHDN Genetic Testing and Counseling Working Group i 2012.Hvordan er prosedyren ved predektiv genetisk testing?
Den genetiske testen bestemmer antallet CAG-repetisjoner i HTT-genet. Testen kan avsløre om du har HS-forlengelsen, men den kan ikke fastslå når symptomene på sykdommen vil begynne, hvor raskt sykdommen vil utvikle seg, eller hvilke symptomer du vil få. Den genetiske testen for HS anses å være nær 100% nøyaktig. Resultatene fra DNA-analysen blir vanligvis dobbeltkontrollert ved å undersøke to separate blodprøver. I tillegg kan blod fra en forelder til den berørte personen (eller, hvis det ikke er tilgjengelig, fra et annet familiemedlem) også bli undersøkt for å bekrefte den opprinnelige diagnosen.Hva er det den genetiske testen undersøker?
Ja. Dette kan oppnås ved hjelp av en moderne diagnostisk prosedyre, kalt preimplantasjonsdiagnostikk (PGD) – som brukes i kombinasjon med in vitro fertilisering (IVF), og involverer screening av embryoer før de settes inn i livmoren. Teknikken sikrer at kun embryoer som arver normale kopier av genet, implanteres i livmoren. Derfor gjør PGD det mulig for et par å få et barn som ikke bærer det forlengede HTT-genet, selv om en av foreldrene bærer HS-mutasjonen – uavhengig av om bæreren er mannen eller kvinnen. I noen land er PGD imidlertid ikke tillatt på grunn av embryobeskyttelseslover, og det er også viktig å merke seg at sjansen for å bli gravid graviditet etter PGD/IVF er lavere, enn hvis man blir gravid på naturlig måte. I enkelte land er det mulig å teste ufødte foster, som er unnfanget på naturlig måte, for så å velge abort når fostrets genetiske status er kjent.Er det mulig for en person, som bærer HS-forlengelsen, å sikre seg at
vedkommende ikke viderefører HS til sine barn?
Prenatal (før fødsel) diagnose er bare mulig når de, som ber om å få det utført, kan vise at saken oppfyller visse medisinske og juridiske kriterier. Disse kriteriene kan variere fra land til land. Det finnes to standardprosedyrer for prenatal diagnose. Den første er en fostervannsprøve, hvor fostervæske som inneholder celler fra fosteret, innhentes via en nål gjennom mors mage, vanligvis etter 14. svangerskapsuke. Den andre er morkakebiopsi, som innebærer innsamling av en vevsprøve fra morkaken og som kan utføres tidligere – mellom 10. og 13. svangerskapsuke. Denne prosedyren innebærer imidlertid en større risiko for fosteret.Kan jeg teste mitt ufødte barn?
Det finnes for øyeblikket ingen terapier som effektivt kan behandle de underliggende årsakene til HS. Basalforskning og klinisk forskning har imidlertid økt vår kunnskap om HS betydelig de seneste årene, og det er nå mange studier på vei, som sikter mot å utsette sykdomsutbrudd eller redusere hastigheten sykdommen utvikler seg. Det finnes allerede behandlinger, som kan lindrer visse symptomer (symptomatiske behandling), og dermed forbedrer pasientens livskvalitet. Disse er delt inn i farmakologiske (legemidler) og ikke-farmakologiske behandlinger.Finnes det behandlinger for HS?
Chorea, bradykinesi, irritabilitet, apati, depresjon, angst og søvnforstyrrelser har alle blitt rapportert å være de mest plagsomme symptomene på HS. Det finnes flere alternativer for å håndtere disse symptomene ved hjelp av legemidler. Mange medisiner kan imidlertid forårsake bivirkninger, og noen kan motvirke den terapeutiske effekten av andre legemidler. Videre kan det samme legemidlet ha forskjellig virkning fra person til person. Behandlingen må derfor tilpasses av en erfaren spesialist på HS, henhold til pasientens symptomer og hans/hennes reaksjoner på de aktuelle legemidlene.Hvilke farmakologiske muligheter finnes for å behandle HS-symptomer?
Ikke-farmakologiske behandlinger (slik som psykoterapi, og kognitiv-, fysio-, tale-, respiratorisk- og ergoterapi) kan forbedre de psykologiske og fysiske symptomene på HS. For eksempel har det blitt rapportert forbedringer i humør, motorisk kontroll, tale, balanse, evne til å svelge og gå etter disse behandlingene. Det er velkjent at mosjon forbedrer både fysisk og mental helse, forbedrer generell trivsel, og mosjon har vist seg å lindre symptomene på depresjon. Det er mer og mer som tyder på at mosjon også kan bidra til å bremse utviklingen av de motoriske symptomene ved HS. For eksempel har noen fysioterapiprogrammer vist seg å forbedre de motoriske symptomer, gang og balanse. EHDNs Physiotherapy Working Group har utgitt et veiledningshefte for fysioterapeuter som arbeider med HS-pasienter.Hvordan kan ikke-farmakologisk behandling hjelpe?
Fordelene med et kosthold rik på vitaminer, co-enzymer og andre komponenter har vært gjenstand for mye diskusjon, men er klinisk aldri blitt bevist. Men, da vekttap er et problem for noen HS-pasienter, spesielt i de senere stadiene av sykdommen, er det viktig å sørge for et sunt kosthold gjennom hele sykdomsforløpet. I de sene sykdomsstadier kan det være nødvendig med en høykalori-diett. Henvisning til en klinisk ernæringsfysiolog kan være nyttig.Kan en spesiell diett lindre HS-symptomene?
Arv & hva er årsaken til HS?
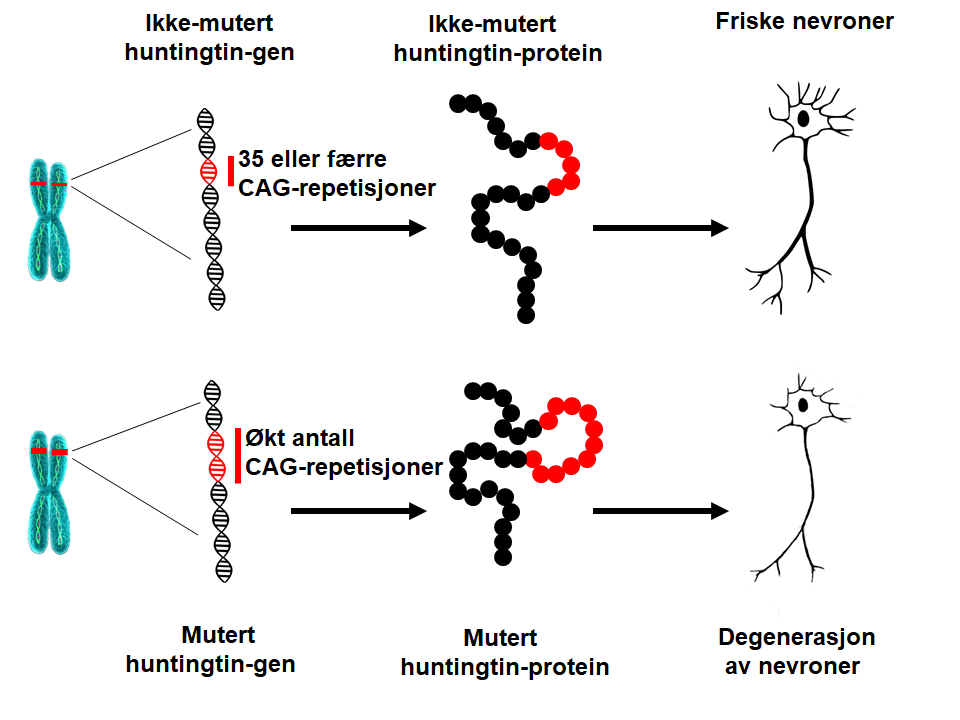
HS skyldes en endring (en forlengelse) i genet (HTT) som koder for et protein kalt huntingtin. Som et resultat av denne forlengelsen, translateres (oversettes) genet til en endret form av proteinet, noe som resulterer i funksjonsfeil og død av nerveceller (nevroner) i bestemte områder av hjernen. De eksakte sykdomsmekanismene er mangesidige og svært komplekse, hvilket er i overenstemmelse med de mange forskjellige funksjonene til huntingtin-proteinet. Forskere jobber med å få en bedre forståelse av de underliggende sykdomsfremkallende mekanismene for å utvikle sykdomsmodifiserende behandlinger.Hva er årsaken til HS?
I 1993 identifiserte forskere mutasjonen som forårsaker HS. HTT-genet er lokalisert på kromosom 4 og koder for et protein kalt huntingtin. Genet inneholder en sekvens av tre nukleotider (de grunnleggende enhetene av DNA), cytosin-adenin-guanin (CAG), som gjentas mange ganger. Denne såkalte trinucleotid-repetisjonen kan variere i lengde. Hvis en person har 40 CAG-repetisjoner eller mer i den ene kopien av HTT-genet, vil han/hun utvikle HS innen normal levetid – det vil si i midten av voksenlivet. Siden repetisjonen som forårsaker HS er tilstede i alle kroppens celler fra unnfangelse, og kan videreføres til etterfølgende generasjoner, er HS en arvelig sykdom.Hva er den underliggende årsaken til HS?
Etter hvert som antallet CAG-repetisjoner øker, blir den delen av DNAet mer ustabil. Dette betyr at antall repetisjoner i denne delen kan øke eller reduseres når den gis videre til neste generasjon. Så lenge antallet CAG repetisjoner i HTT-genet er lavere enn 27, er området stabilt. Hvis antall repetisjoner er mellom 27 og 35 (den såkalte intermediære repetisjonslengden), vil personen ikke selv utvikle HS og genet anses som normalt. CAG- lengder på 27 repetisjoner eller mer er imidlertid ustabile og kan forlenges når de gis videre til neste generasjon, noe som betyr at disse barna har en risiko for å utvikle HS. Personer som har mellom 36 og 39 kan utvikle HS, men hvis de gjør det, er det normalt kun meget sent i livet. Derfor har CAG-repetisjonslengder på mellom 36 og 39 nedsatt penetrans – også kalt gråsonen. Når antallet CAG-repetisjoner er høyere enn 39, vil personen utvikle HS innen normal levetid – oftest i midten av voksenlivet. I sjeldne tilfeller kan CAG-forlengelsen være svært lang, noe som fører til sykdomsutbrudd i tenårene eller i barndommen (juvenil HS). Pasienter som utvikler sykdommen før 10 år, har ofte mer enn 80 CAG-repetisjoner.Hva betyr lengden på HTT CAG-repetisjonen?
CAG-lengde Forårsaker sykdom? Konsekvenser for etterkommere? Navn
Under 27 Nei Ingen Normal lengde
27-35 Nei Repetisjoner på 27 eller mer kan være ustabile og kan forlenges, når de gis videre til etterkommere Intermediær lengde
36-39 Kanskje Ja, etterkommere har 50% sannsynlighet for å arve det forlengede genet Lengde med nedsatt penetrans
40 og flere Ja Ja, etterkommere har 50% sannsynlighet for å arve det forlengede genet Lengde med full penetrans
Gener finnes på kromosomer inne i hver celle i kroppen vår. Et gen er et stykke DNA som inneholder koden for et bestemt protein; DNA transkriberes (oversettes) til messenger RNA (mRNA), som deretter blir oversatt til proteinet. Vanligvis arver alle to kopier av hvert gen – en fra mor og en fra far. HTT-genet, som koder for huntingtin-proteinet, er det viktige i HS. Når et barn arver en forlenget versjon av HTT-genet, så vil også barnet på et tidspunkt utvikle HS. Foreldrene selv kan allerede ha symptomer på sykdommen, eller først utvikle disse senere.Hva er et gen?
Proteiner er store molekyler som består av byggesteiner kalt aminosyrer. Den nøyaktige sekvensen av aminosyrer i et bestemt protein bestemmes av DNA-sekvensen av det tilsvarende genet. Gener fungerer derfor som byggetegninger for cellene – ett sett instruksjoner som forteller cellen hvordan de skal bygge bestemte proteiner. HTT-genet inneholder instruksjoner om hvordan man bygger huntingtin-proteinet. Proteinene er molekylene som gjør nesten alt arbeidet inne i cellene – de utfører et stort antall viktige prosesser, som for eksempel enzymreaksjoner eller strukturell støtte. Hvis et protein fungerer unormalt eller ikke finnes, kan det påvirke cellen og til slutt hele organismen, noe som kan medføre sykdom.Hva er et protein?
Huntingtin-proteinet er et veldig stort protein, som lages eller uttrykkes i varierende grad i hver celle i menneskekroppen; de høyeste nivåene finnes i hjernen. Huntingtin virker å være et veldig viktig protein, fordi musefostre dør hvis ikke proteinet uttrykkes. I den ene enden av huntingtinproteinet er det et område hvor en bestemt aminosyre kalt glutamin gjentas. Denne gjentagelsen, kjent som polyglutaminrepetisjonen, består vanligvis av opptil 35 glutamin-enheter. Hos mennesker som har HS-forlengelsen, finnes det minst 36 gjentagelser, og det er denne polyglutamingjentagelsen som resulterer i et protein som ikke fungerer normalt.Huntingtin-proteinet
HS er en arvelig, dominant sykdom. Dette betyr at en person født med en kopi av det forlengede HTT-genet vil utvikle HS, selv om han/hun også bærer en normal kopi av genet. Sannsynligheten for at en person, som bærer HTT-forlengelsen, gir den videre til sine barn er 50% (forutsatt at vedkommende kun bærer utvidelsen på en av de to kopiene av HTT-genet). Medisinske teknikker kan sikre, at en person, som bærer det forlengede genet kun viderefører det normale HTT-genet til sine barn. På den annen side vil en person som ikke har arvet det forlengede HTT-genet ikke utvikle HS, og hans eller hennes barn vil heller ikke være i fare for å utvikle sykdommen. HS-forlengelsen kan ikke hoppe over en generasjon. Det kan imidlertid skje at en bærer av det forlengede HTT-genet dør før symptomene oppstår, og at vedkommendes barn derfor ikke skjønner at de er i fare for å utvikle sykdommen.Hvordan videreføres HS?
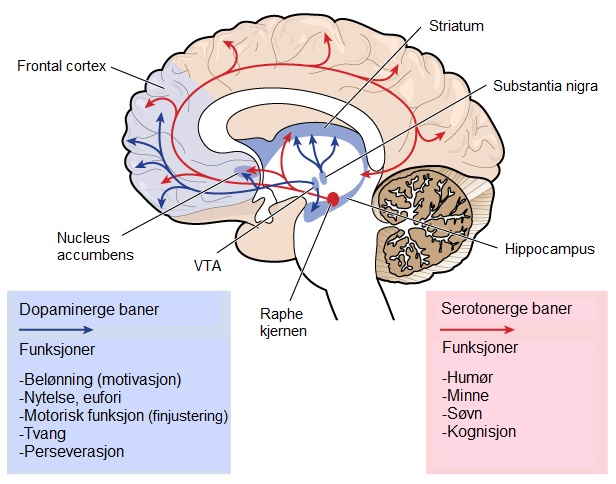
Visse funksjoner i hjernen, som evnen til å bevege seg, tenke og snakke, svekkes gradvis ved HS, i takt med at viktige nerveceller skades og dør. Den delen av hjernen som blir hardest rammet av HS er striatum, som er en del av de basale ganglier og ligger dypt i hjernens sentrale region. Striatum er primært involvert i planlegging og styring av bevegelser, men også i mange andre prosesser, inkludert kognisjon og følelser. Ettersom HS utvikler seg, påvirkes også cortex (den ytterste, rynkete delen av hjernen), noe som bidrar til de kognitive symptomene. Over tid forårsaker HS generell atrofi av hele hjernen, noe som svekker individets generelle funksjonsnivå.Hjerneområder som er involvert i HS
HS i hverdagen
Å teste positivt for HS-forlengelsen kan påvirke mange forskjellige aspekter av en persons liv, inkludert beslutningen om hvorvidt man skal å få barn eller ikke, hvordan man skal planlegge fremtiden, om man må revurdering tidligere prioriteringer, hvordan man skal bo og hvorvidt man skal informere andre familiemedlemmer om at de også kan være i fare for å ha arvet sykdommen. Spesielt unge voksne må tenke over hvilke konsekvenser et positivt testresultat vil få for utdanning og jobb. Etter hvert som sykdommen utvikler seg, påvirker det gradvis en persons evne til å leve uavhengig av andre. Arbeid, sosialt liv og daglige aktiviteter blir mer og mer problematiske, og pasienter blir i stigende grad avhengige av hjelp fra slektninger og helsepersonell. Lokale pasientforeninger og spesialisthelsetjenesten kan kontaktes, hvis man trenger hjelp.Hvordan påvirker HS hverdagen?
Effektive strategier for å håndtere HS må tilpasses det enkelte individ, så man kan ta høyde for sykdomsstadiet og familiekonteksten. HS utvikler seg veldig sakte, slik at det generelt er tid til å tilpasse seg de endringene som sykdommen medfører. For pleiepersonell og nærmeste familie kan en bedre forståelse av de atferdsmessige og kognitive svekkelsene, som er forbundet med sykdommen, kanskje hjelpe dem å finne strategier for å imøtekomme forandringene og hjelpe dem med å opprettholde et godt forhold til personen med HS. Nyttige råd og veiledning kan fås ved å kontakte HS-spesialister og pasientforeninger.Finnes det strategier for hvordan man bedre håndterer å leve med HS?
Dersom du kikker på denne siden, finner du en liste over “language area coordinators” som kan hjelpe deg. Alternativt kan du benytte dette kontaktskjemaet.Hvordan kontakter jeg EHDN?
For å få en time hos en HS-spesialist må du ha en henvisning fra din fastlege. Hvis du er i tvil, kan du kontakte din lokale EHDN Language coodinator som vil hjelpe deg.Hvordan får jeg en avtale med en HS-spesialist?
Uavhegig hjelp kan fås ved å kontakte pasientforeningen i ditt land.Er det er mulighet for å få snakke med en spesialist utan å gå via
sykehussystemet?
EHDN spiller en nøkkelrolle i den verdensomspennende kliniske studien Enroll-HD. Dette er en observasjonsstudie som ikke involverer intervensjoner. Det betyr at den ikke prøver ut ny eksperimentell behandling per se. Deltakerne vurderes klinisk en gang i året, og noen kan være kvalifisert til å delta i kliniske studier av symptomatiske eller sykdomsmodifiserende behandlinger hvis/når disse blir tilgjengelige. Enroll-HD er tilgjengelig på mange spesialiserte HS klinikker/avdelinger rundt om i verden. For å finne ut om det finnes et Enroll-HD senter i nærheten av deg kan du se her eller henvende deg til din lokale EHDN language coordinator som vil kunne informere deg om forskningsaktiviteter i din region. Din lokal pasientforening vil kunne gi deg generell informasjon om deltakelse i forskning. For mer informasjon om HS-forskning, se her eller besøk nettsiden HDBuzz, som inneholder HS-forskningsnyheter skrevet av HS-forskere på et språk alle kan forstå og som er oversatt til mange ulike språk.Hvordan kan jeg bli involvert i HS-forskning?
Ja, en rekke pasientforeninger gir støtte til enkeltpersoner og familier som er rammet av HS. Disse foreningene kan kontaktes direkte. Den europeiske Huntingtonforeningen (EHA) fører en liste over pasientforeninger som kan være til nytte for deg.Finnes det støttegrupper som er spesialisert innen HS?
Dersom du har spørsmål er du velkommen til å ta kontakt med din EHDN koordinator eller din lokale pasientforening.