Historia HD
Nazwa choroby wzięła się od George’a Huntingtona, amerykańskiego lekarza, który opisał ją w 1872 roku. Jego opis opierał się na obserwacjach rodzin, żyjących w wiosce East Hampton, na wyspie Long Island, niedaleko Nowwego Jorku, gdzie dr Huntington żył i pracował. Dawniej chorobę nazywano pląsawicą Huntingtona lub tańcem Św Wita.
Objawy & Postęp choroby
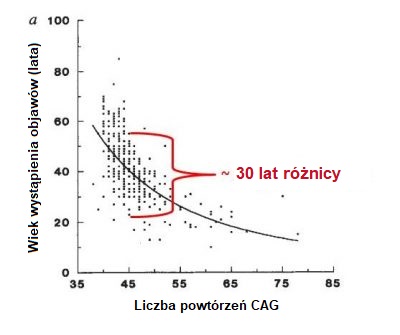
HD jest rzadką chorobą, która dotyka od 5 do 10 na 100000 osób w populacji europejskiej. Podobna częstość występowania dotyczy tych krajów, których mieszkańcy są pochodzenia europejskiego, np. USA. HD jest schorzeniem rzadkim w Azji i Afryce, gdzie występuje u ok 1 na 100000 osób. Ryzyko odziedziczenia nieprawidłowego genu i zachorowania na HD jest równe dla kobiet i mężczyzn. HD cechuje współwystępowanie objawów ruchowych, zaburzeń zachowania (np. zaburzeń nastroju) i poznawczych (np. problemów z rozumieniem). Mogą występować również i inne objawy. Objawy HD cechuje duża zmienność w zakresie ciężkości, początku występowania i szybkości progresji – zmienność ta obserwowana jest nawet u członków tej samej rodziny. U jednej osoby mogą występować wyraźne zaburzenia ruchowe i jednocześnie łagodne objawy poznawcze oraz psychiatryczne, podczas gdy inny pacjent może cierpieć z powodu depresji i zaburzeń lękowych na wiele lat przed pojawieniem się objawów motorycznych. Początek HD jest często opisywany jako podstępny, ponieważ bywa trudno określić datę pojawienia się objawów. Większość osób będących nosicielami mutacji warunkującej HD rozwija objawy choroby pomiędzy 35 a 55 rokiem życia. U ok 10% nosicieli objawy pojawiają się przed 20 rokiem życia (postać młodzieńcza choroby – Juvenile HD), także u 10% początek objawów może nastąpić po 55 roku życia. Ogólnie objawy pojawiają sie stopniowo i przez wiele lat mogą pozostać nierozpoznane. Średnio czas trwania choroby wynosi 15-20 lat od chwili postawienia diagnozy, ale czas ten jest zmienny i zależy od jakości opieku nad chorymi. Czynniki wpływające na czas wystąpienia objawów sa bardzo złożone i obecnie poddawane intensywnym badaniom. Obserwacja dużych grup pacjentów pozwoliła znaleźć uczonym zależność pomiędzy liczbą powtórzeń trójki nukleotydów a wiekiem wystąpienia objawów choroby (Rycina poniżej). Stwierdzono, że generalnie im większa liczba powtórzeń CAG, tym szybszy początek objawów (rycina poniżej). Co jest jednak ważne, dla każdej liczby powtórzeń CAG zmienność wieku wystąpienia początku choroby może wynosić nawet 30 lat, jak pokazano to na wykresie poniżej. Przyczyną prawdopodobnie jest wpływ innych genów (tak zwanych genetycznych modyfikatorów choroby), oraz czynników środowiskowych takich jak styl życia i dieta. Biorąc to wszystko pod uwagę, jest bardzo trudno dokładnie przewidzieć wiek zachorowania u poszczególnych osób – nosicieli genu powodującego chorobę. Klasyfikacja opracowania przez neurologa i specjalistę od HD Irę Shoulsona z Georgetown University w USA dzieli przebieg choroby na pięć stadiów: Gdy choroba rozpoczyna się we wczesnym okresie życia (przed osiągnięciem wieku 20 lat), spowolnienie ruchowe (bradykinezja) oraz dystonia są zwykle bardziej nasilone od ruchów mimowolnych (pląsawicy). Wczesnymi objawami młodzieńczej postaci choroby są też zaburzenia zachowania, problemy z mową i uczeniem się, oraz narastające trudności w szkole. Czasami występują napady padaczkowe, które są częstsze u pacjentów młodszych. Ogólnie postęp młodzieńczej postaci choroby jest szybszy niż postaci dorosłych. Przy późnym początku choroby pląsawica stanowi objaw dominujący nad bradykinezją i dystonią. W takich wypadkach jest trudniej zebrać wywiad rodzinny, gdyż często rodzice pacjenta już nie żyją, a jeśli zmarli wcześnie, mogło u nich nie dojść do pojawienia się objawów choroby. Przyczyną śmierci nie jest bezpośrednio choroba, tylko powikłania związane z ogólnym osłabieniem organizmu. Należą do nich: zapalenie płuc (będące przyczyną śmierci 1/3 pacjentów z HD) , zachłyśnięcie się, atak serca, uraz głowy jako konsekwencja upadku, zaburzenia odżywiania. HD wiąże się z wysokim ryzykiem samobójstwa, które odpowiada za ok 7% przypadków śmierci.Jak często zdarza się HD?
Jakie są objawy HD?
Najczęstszymi psychiatrycznymi objawami HD są apatia, zaburzenia lękowe, depresja, drażliwość, wybuchy gniewu, impulsywność, zaburzenia obsesyjno-kompulsyjne, zaburzenia snu i wycofanie się z życia społecznego. Nieco rzadziej występują mania i objawy psychotyczne – urojenia (fałszywe przekonania) oraz halucynacje (widzenie, słyszenie lub odczuwanie nieistniejących bodźców). Często występują myśli samobójcze, zwłaszcza we wczesnych stadiach choroby. Zaburzenia psychiatryczne odbierane są przez wielu pacjentów i ich opiekunów jako bardziej dokuczliwe niż objawy ruchowe i poznawcze.
Cechą HD jest postępujące upośledzenie rozumienia, wnioskowania, pamięci i zdolności do oceny. Do objawów poznawczych należą spowolnienie myślenia, problemy z koncentracją, organizacją, planowaniem, podejmowaniem decyzji, odpowiadaniem na pytania. Do kłopotów z pamięcią krótkoterminową i zdolnością odpowiadania na pytania dołączają się problemy z uczeniem się i rozumieniem nowych informacji.
W przebiegu HD może wystąpić wiele innych objawów takich jak utrata apetytu, utrata wagi, zaburzenia samooceny, zaburzenia seksualne, problemy z funkcjonowaniem zwieraczy.Kiedy pojawiają się objawy HD?
Co wpływa na czas wystąpienia objawów?
Jakie są stadia zaawansowania choroby?
Czy objawy postaci młodzieńczej choroby Huntingtona różnią się od
postaci dorosłych?
Jakie są objawy HD, gdy choroba rozpoczyna się w późnym
okresie życia?
Przyczyny śmierci
Diagnostyka & leczenie
Diagnostyka HD opiera się na badaniach lekarskich oraz na teście genetycznym. Kliniczna diagnostyka opiera się na wywiadzie lekarskim i rodzinnym, oraz na specjalnych skalach klinicznych, które pozwalają ocenić ciężkość i czestość występowania objawów. Wyniki badania lekarskiego zwykle zostają uzupełnione badaniem genetycznym oceniającym liczbe powtórzeń CAG w genie HTT (diagnostyczne badanie genetyczne). Jeśli u pacjenta nie występują objawy choroby badanie genetyczne przedobjawowe określa, czy jest on nosicielem mutacji.W jaki sposób diagnozowana jest choroba Huntingtona?
Narzędzia oceny klinicznej, stosowane w diagnostyce HD i ocenie różnych jej objawów różnią się pomiędzy ośrodkami i krajami. Najczęściej wykorzystywana jest tzw skala UHDRS (Unified Huntington Disease Rating Scale), która zawiera cześci poświęcone ocenie objawów ruchowych, behawioralnych, poznawczych i stanu funkcjonalnego. Dodatkowo kwestionariusz Problems Behaviour Assessment (PBA) stosowany jest w ocenie częstości występowania i stopnia nasilenia zaburzeń zachowania (takich jak depresja, apatia, drażliwość). Wiele testów, takich jak Mini Mental State Examination (MMSE) i Mattis Dementia Rating Scale) wykorzystywanych jest do uzupełnienia oceny zaburzeń poznawczych.Jakie narzędzia oceny klinicznej stosowane są w diagnostyce HD?
Życie ze świadomością nosicielstwa choroby Huntingtona może być bardzo trudne. Możesz jednak stwierdzić, że wolisz uzyskać informację na temat wyniku swojego stanu genetycznego. W takiej sytuacji bardzo zalecane jest poradnictwo genetyczne oraz opieka psychologiczna, które pozwolą ci omówić z lekarzem i terapeutą istotne kwestie oraz podjąć życiowe decyzje. Ogólnie, badanie genetyczne nie jest zalecane osobom poniżej 18-go roku zycia. Wydaje się, że osoby w wieku dorosłym mają wystarczającą dojrzałość aby zmierzyć się z faktem nosicielstwa. W niektórych wypadkach warto jest przeprowadzić potwierdzające badanie genetyczne u dzieci – wówczas, gdy zaobserwowano u nich objawy sugerujące postać młodzieńczą choroby, lub u kobiet poniżej 18-go roku życia, będących w ciąży. Jeśli podejmiesz decyzję o badaniu genetycznym, pobrana zostanie z żyły na ramieniu mała ilość krwi, z której wyizolowane zostanie DNA. W zależności od lokalnego laboratorium wynik otrzymasz po 2-8 tygodniach. Wytyczne dotychczące zasad przedobjawowego badania genetycznego zostały uaktualnione przez Grupę Roboczą do Spraw Badań Genetycznych i Poradnictwa działającą przy EHDN w roku 2012.W jaki sposób przeprowadza się przedobjawowe badanie genetyczne?
Badanie genetyczne określa liczbe powtórzeń CAG w genie HTT. Test wykazuje czy w genie znajduje się zmiana odpowiadającająca za wystąpienie choroby, nie odpowiada jednak na pytanie, kiedy pojawią się objawy, jak szybko będą postępować, lub jakiego rodzaju będą. Dokładność testu jest bliska 100%. Wyniki badania DNA są sprawdzane badaniem dwóch próbek krwi. Dodatkowo krew rodzica lub innego członka rodziny moze być również zbadana aby potwierdzić diagnozę.Co wykrywa badanie genetyczne?
Tak. W tym celu wykonuje się nowoczesną procedurę, która nazywa się genetyczną diagnostyką preimplantacyjną (Preimplantation Genetic Diagnosis – PGD), nazywaną czasem też screeningiem embrionów. Procedurę tą stosuje się w połączeniu z zapłodnieniem pozaustrojowym, i polega na przeprowadzeniu badania genetycznego embrionów przed ich umieszczeniem w macicy. Technika ta pozwala uzyskać pewność, że tylko embriony zawierające prawidłowy gen HTT zostana umieszczone w macicy. PEG umożlwia więc kobietom zajście w ciążę i posiadanie zdrowego dziecka, niezależnie od tego, czy nosicielem mutacji jest kobieta, czy jej partner. Diagnostyka preimplantacyjna w niektórych krajach jest zabroniona, należy również pamiętać, że szanse na zajście w ciążę przy zapłodnieniu pozaustrojowym są niższe niż w wypadku naturalnego poczęcia. W niektórych krajach możliwa jest diagnostyka genetyczna płodu i podjęcie decyzji o aborcji po uzyskaniu informacji o nosicielstwie.Czy osoba będąca nosicielem mutacji HD może upewnić się, że nie
przekazała jej swojemu dziecku?
Diagnostyka prenatalna (przedurodzeniowa) możliwa jest wówczas, gdy osoby, które o nią proszą spełnią odpowiednie kryteria medyczne i prawne, które zależą od kraju zamieszkania. Stosowane są dwie standardowe procedury diagnostyki prenatalnej: Pierwszą jest amniocenteza (zwana także badaniem płynu owodniowego). W czasie tej procedury płyn owodniowy pobierany jest przy pomocy igły, którą nakłuwa się brzuch matki. Badanie to wykonywane jest zwykle po 14 tygodniu ciąży. Druga procedura to badanie kosmków owodniowych, która polega na biopsji tkanki łożyska i może być przeprowadzona wcześniej – pomiędzy 10-a 13 tygodniem ciąży. Jest to badanie nieco bardziej ryzykowne dla płodu.Czy mogę zbadać moje nienarodzone dziecko?
W chwili obecnej nie są jeszcze dostępne sposoby leczenia przyczyn choroby Huntingtona. Dzięki badaniom podstawowym i klinicznym nasza uległa znacznemu zwiększeniu, i trwają obecnie liczne badania, których celem jest identyfikacja leków mogących spowolnić postęp choroby, lub odsunąć w czasie jej początek. Leczenie dostępne aktualnie jest w stanie złagodzić niektóre objawy choroby (leczenie objawowe) i w ten sposób poprawić jakość życia pacjentów. Metody leczenia dzielą się na farmakologiczne i niefarmakologiczne.Czy można leczyć chorobe Huntingtona?
Pląsawica, spowolnienie ruchowe, drażliwość, apatia, depresja, lęk i zaburzenia snu uważane są na najbardziej dokuczliwe objawy HD. Istnieją sposoby łagodzenia tych objawów przy pomocy leków. Wiele leków niestety wywiera działania niepożądane, ponadto gdy stosowanych jest kilka leków, ich działania mogą się nawzajem znosić. Co więcej, ten sam lek może wywierać różne skutki u różnych pacjentów. Leczenie musi być dostosowane do pacjenta przez specjalistę z dużym doświadczeniem w leczeniu HD, na podstawie objawów chorego i jego odpowiedzi na leczenie.Jakie są możliwości farmakologicznego leczenia objawów HD?
Metody niefarmakologiczne, takie jak psychoterapia, fizykoterapia, zajęcia z logopedą, terapia zajęciowa, ćwiczenia oddechowe mogą zmniejszyć nasilenie psychicznych i fizycznych objawów HD. Na przykład, obserwowano redukcję nasilenia zaburzeń nastroju, mowy, problemów z poruszaniem się, równowagą, połykaniem pokarmu i chodem. Wiadomo, że ćwiczenia pozytywnie wpływają zarówno na zdrowie fizyczne jak i psychiczne, poprawiają samopoczucie oraz mogą łagodzić objawy depresji. Coraz więcej dowodów wskazuje, że ćwiczenia fizyczne spowalniają postęp niesprawności ruchowej w HD. Na przykład wykazano, że niektóre programy fizykoterapeutyczne mogą redukować zaburzenia ruchowe, poprawiać sprawność chodu i równowagę. Grupa Robocza do Spraw Fizykoterapii opracowała wytyczne dla fizykoterapeutów pracujących z pacjentami z HD.Czy niefarmakologiczne sposoby leczenia mogą pomóc pacjentom?
Skuteczność diet bogatych w witaminy, koenzymy i inne składniki jest przedmiotem wielu rozważań, nie udowodniono jednak do tej pory ich pozytywnego wpływu na przebieg HD. W późniejszych stadiach choroby duży problem stanowi utrata wagi i wówczas niezwykle istotne jest zapewnienie zdrowej i zbilansowanej diety. Konieczne może być stosowanie diety wysokokalorycznej. Pomocna może być wowczas porada dietetyka.Czy dieta może złagodzic objawy HD?
Inheritance & Co powoduje HD?
HD jest powodowana zmianą (wydłużeniem) w genie (HTT) który koduje białko zwane huntingtyna. Jako wynik tego wydłużenia gen produkuje zmienione białko, co skutkuje zaburzeniami funkcji i śmiercią komórek (neuronów) w określonych obszarach mózgu. W szczegółach patomechanizm choroby jest wieloaspektowy i bardzo złożony, co wynika ze złożoności funkcji białka huntingtyny. Naukowcy pracują nad lepszym zrozumieniem czynników wywołujących chorobę aby stworzyć terapie modyfikującą jej przebieg.Co powoduje HD?
Naukowcy w 1993 roku wykryli mutację powodującą HD. Gen HTT znajduje się na chromosomie 4 i koduje białko zwane huntingtyną. Ten gen zawiera trójnkleotydową sekwencję (podstawowe składniki DNA) cytozyna-adenina-guanina (CAG), która jest wielokrotnie powtórzona. Te tak zwane trójnukleotydowe powtórzenia mogą się różnić długością. jeśli ktoś ma 40 powtórzeń CAG lub więcej w jednej kopii genu HTT wówczas w ciągu normalnej długości życia zachoruje na HD – zwykle w wieku średnim-dorosłym. Ponieważ wydłużenie w genie HTT powodujące HD jest obecne w ciele od poczęcia i może być przeniesione na kolejne pokolenia toteż HD jest chorobą dziedziczną.Jaka przyczyna leży u podłoża HD?
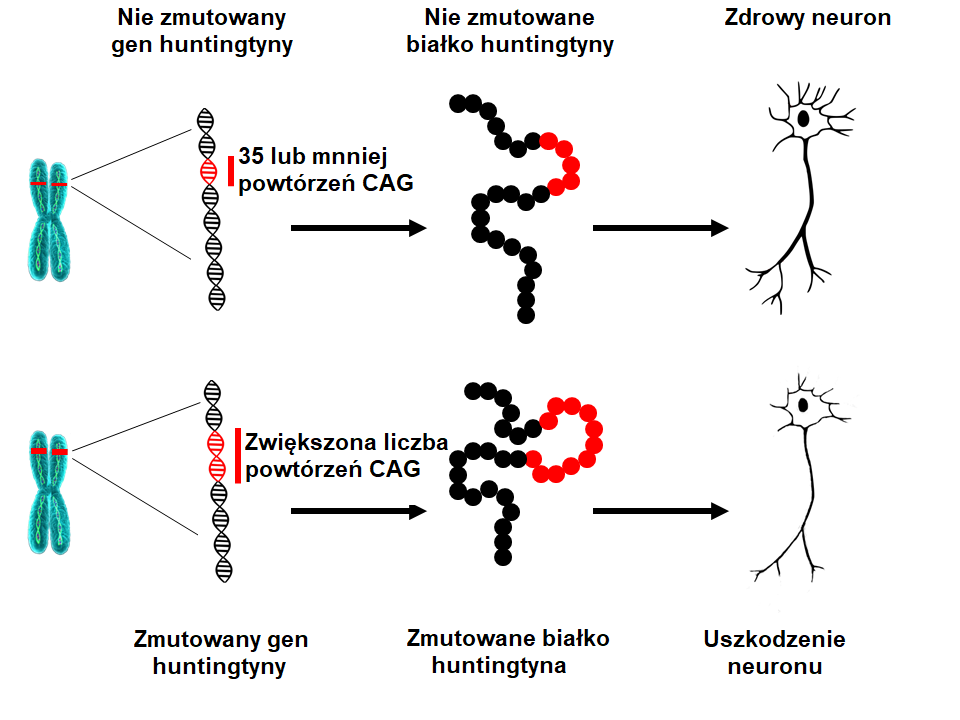
Gdy liczba powtórzeń CAG wzrasta wówczas określona część DNA staje się niestabilna. Niestabilność oznacza, że liczba powtórzeń CAG w tej części DNA może się jeszcze zwiększyć lub zmniejszyć gdy gen jest przekazywany potomkowi. Tak długo jak liczba powtórzeń CAG w genie HTT jest mniejsza niż 27 jest on stabilny. Gdy liczba powtórzeń wynosi pomiędzy 27 a 35 (tak zwana pośrednia liczba powtórzeń), wówczas osoba taka nie zachoruje na HD, a wspomniana część DNA jest uważana za prawidłową, ale gdy powtórzeń CAG jest więcej niż 27 wówczas staje się ona niestabilna i podatna na wydłużenie gdy przechodzi do potomka. Oznacza to że potomek jest w grupie ryzyka zachorowania na HD. Osoby które posiadają pomiędzy 36 a 39 powtórzeń mogą zachorować na HD, ale dopiero w wieku podeszłym, o ile w ogóle zachorują na HD. Te wartości powtórzeń są określane mianem zakresu powtórzeń o obniżonej penetracji. Gdy liczba powtórzeń CAG jest wyższa niż 39 wówczas taka osoba zachoruje na HD w ciągu normalnej długości życia – najczęściej w wieku średnim-dorosłym. W rzadkich przypadkach liczba powtórzeń CAG może być szczególnie duża, prowadząc do zachorowania w wieku młodzieńczym lub w dzieciństwie (postać młodocianych HD). Pacjenci którzy chorują w wieku młodszym niż 10 lat często mają ponad 80 powtórzeń CAG.Co oznacza liczba powtórzeń CAG w genie HTT
Liczba powtórzeń CAG Czy wsytąpi choroba? Czy istnieje ryzyko dla potomstwa? Nazwa
Poniżej 27 Nie Żadne Normalna liczna powtórzeń
27-35 Nie Liczba 27 i więcej powtórzeń może być niestabilna i może ulec zwiększeniu , gdy zostanie przekazana potomstwu. Pośrednia liczba powtórzeń
36-39 Możliwe Tak, ryzyko przekazania potomstwu zmutowanego genu wynosi 50%. Liczba powtórzeń o zredukowanej penetracji
40 i powyżej Tak Tak, ryzyko przekazania potomstwu zmutowanego genu wynosi 50%. Liczba powtórzeń o pełnej penetracji
Geny znajdują się na chromosomach w każdej komórce naszego ciała. Gen jest obszarem DNA w którym zawarty jest kod określonego białka; DNA podlega transkrypcji na informacyjne RNA (mRNA), które następnie podlega procesowi translacji tworząc białko. Najczęściej każdy dziedziczy dwie kopie każdego genu – jedną od matki, druga od ojca. Ważny dla HD jest gen HTT który koduje białko huntingtynę. Gdy osoba odziedziczy wydłużona wersje genu HTT wówczas zachoruje na HD. Rodzic może być już chory na HD lub zachoruje w późniejszym wieku.What is a gene?
Białka to duże molekuły zbudowane z cząsteczek zwanych aminokwasami. Kolejność aminokwasów w białku jest określona przez DNA genu kodującego to białko. Geny zatem działają jak plan – zestaw instrukcji dla komórki informujących jak mają być zbudowane białka. Gen HTT zawiera informacje jak zbudować białko huntingtynę. Białka są molekułami wykonującymi prace wewnątrz komórki – wykonują wiele istotnych zadań takich jak reakcje enzymatyczne lub utrzymanie struktury. Jeśli białko działa nieprawidłowo lub go brakuje z powodu wydłużenia genu który je koduje, wówczas może to uszkodzić komórkę i ostatecznie cały organizm, może to doprowadzić do zachorowania.Co to jest białko?
Huntingtyna jest bardzo dużym białkiem które jest „produkowane” w różnych ilościach w każdej komórce ludzkiego ciała; największe jej ilości znajdują się w mózgu. Huntingtyna wydaje się być bardzo ważnym białkiem ponieważ jej brak prowadzi do śmierci mysich zarodków. Na jednym końcu białka huntingtyny znajduje się odcinek zbudowany z powtórzeń jednego aminokwasu nazywanego glutaminą. Ta charakterystyczna cecha znana jako powtórzenia poliglutaminowe normalnie składa się z nie więcej niż 35 jednostek glutaminy. Osoby które są nosicielami mutacji warunkującej HD posiadają przynajmniej 36 powtórzeń co stanowi wydłużenie ciągu poliglutaminowego skutkującego zaburzeniem funkcji tego białka.Białko huntingtyna
Nosiciel mutacji warunkującej HD, niezależnie czy jest objawowy czy nie, może przenieść na potomka prawidłowa lub zmutowaną kopie genu z 50% prawdopodobieństwem każdej z tych możliwości (zakładając ze posiada mutację tylko jednej kopii genu HTT). Technologie medyczne mogą zapewnić że przeniesiona będzie tylko prawidłowa kopia genu HTT na potomka osoby z mutacją. Z drugiej strony osoba która nie odziedziczyła zmutowanego genu HTT nie zachoruje a jej dzieci również nie będą w grupie ryzyka zachorowania. Mutacja warunkująca HD nie przeskakuje pokolenia. Może się jednak zdążyć że osoba która jest nosicielem mutacji umrze przed osiągnięciem wieku w którym rozwinęłyby się objawy i w ten sposób dzieci tej osoby nie są świadome iż znajdują się w grupie ryzyka zachorowania na tę chorobę.Jak przenosi się HD?
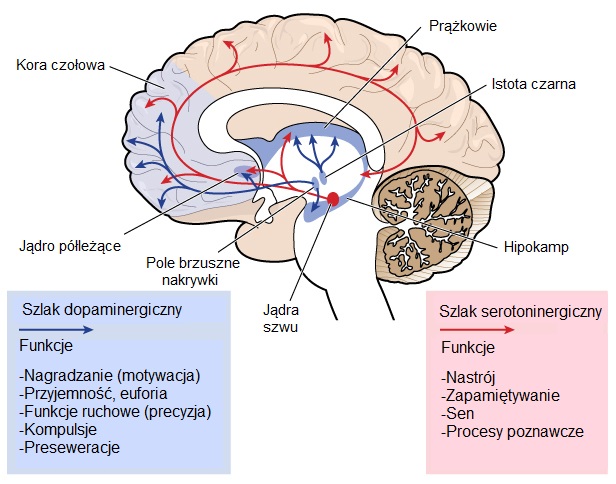
Poszczególne funkcje mózgu takie jak zdolność do poruszania się, myślenia i rozmawiania stopniowo pogarszają się w trakcie trwania HD ponieważ kluczowe dla tych zdolności komórki nerwowe ulegają uszkodzeniu i umierają. Prążkowe, które wchodzi w skład jader podstawy i znajduje się głęboko w środkowym obszarze mózgu, jest częścią mózgu który jest najbardziej dotknięty przez proces chorobowy HD. Prążkowe jest istotne głównie dla planowania i kontrolowania ruchów ale bierze udział również w innych procesach jak poznanie i emocje. Ponieważ postęp HD dotyka również kory (najbardziej zewnętrzna pomarszczona część mózgu), toteż pogarsza zdolności poznawcze. Najogólniej HD powoduje zanik całego mózgu w czasie doprowadzając do obniżenia ogólnych zdolności funkcjonowania.Obszary mózgu objęte HD
HD w życiu codziennym
Pozytywny wynik testu genetycznego w kierunku HD może zaburzyć wiele aspektów życia osobistego takich jak decyzja o posiadaniu potomstwa, planowanie przyszłości, ustalenie priorytetów, negocjowanie odpowiedniego miejsca zamieszkania, lub informowanie innych członów rodziny iż mogą być w grupie ryzyka zachorowania na HD. Młodzi dorośli w szczególności mogą potrzebować przemyśleć implikacje pozytywnego wyniku badania genetycznego w aspekcie ich kształcenia, szkolenia i zatrudnienia. Ponieważ postęp choroby stopniowo zaburza zdolność do samodzielnego życia. Praca, życie społeczne i czynności dnia codziennego staja się problemem i pacjenci staja się coraz bardziej zależni od grupy wsparcia pacjentów i ośrodki kliniczne są dostępne i służą pomocy w każdej chwili.Jak HD wpływa na codzienne życie
Efektywne strategie radzenia sobie z HD muszą być spersonalizowane i dotyczyć chorej osoby, stopnia zaawansowania choroby i rodziny pacjenta. HD rozwija się bardzo powoli dlatego jest czas na dostosowanie się do zmian które ona przynosi. Dla opiekunów i bliskich lepsze zrozumienie zaburzeń zachowania i zdolności poznawczych związanych z chorobą może pomóc w rozwoju strategii akceptacji tych zmian i utrzymania dobrych relacji z chorą osoba. Pomocne informacje i porady są dostępne uspecjalistów zajmujących się HD i grup wsparcia pacjentów.Czy są strategie radzenia sobie z HD?
Jak się skontaktować z EHDN?
Może Pani/Pan umówić się na wizytę po otrzymaniu skierowania od lekarza rodzinnego lub kontaktując się z lokalnym koordynatorem dla strefy językowej EHDN.Jak umówić się do specjalisty?
Niezależne porady dotyczące HD można uzyskać grupy wsparcia pacjentów w Pani/Pana kraju.Czy jest jakiś sposób żeby porozmawiać ze specjalistą bez wybierania
się do kliniki?
EHDN gra kluczową rolę w prowadzeniu badania obserwacyjnego Enroll-HD. Jest to badanie obserwacyjne zatem nie ma w nim żadnej interwencji. To oznacza że nie testuje się żadnej eksperymentalnej terapii w tym badaniu. Uczestnicy przechodzą badania lekarskie w trakcie corocznych wizyt i mogą zostać zaproszeni do badań klinicznych nad leczeniem objawowym lub modyfikujących przebieg choroby gdy one staną się dostępne. Enroll-HD jest dostępne w wielu ośrodkach na całym świecie. Żeby znaleźć ośrodek w pobliżu miejsca zamieszkania proszę zajrzeć tutaj lub zapytać koordynatora Państwa strefy językowej EHDN który będzie w stanie poinformować Państwa o badaniach w Państwa regionie. Państwa grupa wsparcia pacjentów będzie w stanie podać Państwu ogólne informacje na temat uczestnictwa w badaniach. Aby uzyskać więcej informacji na temat badań nad HD proszę zajrzeć tutaj lub odwiedzić stronę internetową HDBuzz gdzie badania są opisane przez naukowców przystępnym jeżykiem i przetłumaczone na wiele języków.nd translated into most languages.Jak mogę się włączyć do badań nad HD?
Tak, liczne grupy wsparcia pacjentów niosą pomoc pojedynczym osobom oraz rodzinom dotkniętym HD. Można z nimi nawiązać kontakt poprzez lekarza rodzinnego albo specjalistę HD albo bezpośrednio się z nimi skontaktować. Europejskie Stowarzyszenia Choroby Huntingtona (EHA) posiada listę grup wsparcia która może być Pani/Panu przydatna.Czy są grupy wsparcia specjalizujące się w HD?