História da DH
A DH deve o seu nome a George Huntington, um médico americano que descreveu a doença em 1872. A sua descrição baseou-se na observação de famílias afectadas pela DH, provenientes da aldeia de East Hampton, Long Island, em Nova Iorque (EUA), onde o Dr. Huntington viveu e trabalhou. A DH era conhecida como coreia de Huntington e previamente como dança de São Vito.
Sintomas & progressão da doença
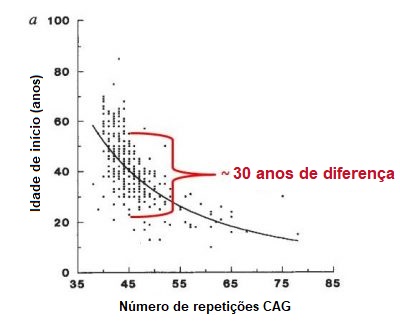
A DH é uma doença rara que afecta 5 a 10 pessoas por cada 100,000 pessoas na população Europeia. Foi encontrada uma prevalência semelhante em países cujas populações têm maioritariamente ascendência europeia, como é o caso dos EUA. A DH é menos comum em países Asiáticos e Africanos, onde a prevalência se estima ser de 1 pessoa em cada 100,000. Homens e mulheres têm probabilidades iguais de herdar a expansão da DH e de desenvolver a doença. A DH é caracterizada pela combinação de alterações motoras (movimento), comportamentais (humor, por exemplo) e cognitivas (como a compreensão), embora se possam também reportar outros sintomas. Os sintomas da DH podem variar em gravidade, idade de início e velocidade de progresssão de indivíduo para indivíduo – mesmo entre membros da mesma família. Uma pessoa pode apresentar uma alteração do movimento muito clara mas discretos sintomas comportamentais e deteriorização cognitiva ligeira, enquanto outra pessoa pode sofrer de depressão e ansiedade durante anos antes de qualquer alteração do movimento. O início da DH é descrito como sendo “insidioso”, visto ser frequentemente dificil determinar uma data concreta de começo da doença. A maioria dos indivíduos portadores da expansão da DH desenvolvem sintomas na meia-idade adulta, entre os 35 e os 55 anos. Aproximadamente 10% apresentam sintomas antes dos 20 anos (têm DH juvenil) e outros 10% após os 55 anos. Em geral, a DH desenvolve-se de forma gradual, podendo passar anos sem ser diagnosticada. Em média, a duração da doença é de 15 a 20 anos após o diagnóstico, existindo variações entre os indivíduos e podendo também depender da qualidade dos cuidados que o doente recebe. Os determinantes da idade de início são complexos e assunto de investigações em curso. Quando se avaliam grandes grupos de doentes com DH, os investigadores encontram uma correlação entre o número de repetições do trinucleótido e a idade de início dos sintomas (figura abaixo). Isto significa que, em geral, quanto maior o número de repetições CAG, mais cedo aparecem os sintomas (figura abaixo). Contudo, por cada número de repetições de CAG, a variação na idade do início dos sintomas pode ir até 30 anos, como demonstrado na figura abaixo. Este facto deve-se, provavelmente, ao efeito de outros genes para além do gene HTT (chamados modificadores genéticos), e a fatores ambientais como o estilo de vida ou a dieta. Tendo tudo isto em conta, é muito difícil prever com precisão a idade em que irão aparecer os sintomas numa pessoa com a expansão da DH. De acordo com a classificação desenvolvida pelo neurologista e especialista em DH Ira Shoulson, da Universidade de Georgetown nos EUA, a progressão da DH pode ser dividida em cinco estadios: Quando a DH começa cedo (antes dos 20 anos de idade), os movimentos involuntários (coreia) são um sintoma menos proeminente que a lentidão do movimento (bradicinésia) e a rigidez (distonia). Características precoces da DH juvenil incluem alterações significativas do comportamento, dificuldades na aprendizagem e fala, e declínio no desempenho escolar. Crises epiléticas são ocasionalmente reportadas, sendo mais comuns em doentes mais novos. Na sua generalidade, a forma juvenil da doença progride mais rapidamente que a sua forma adulta. Quando a DH se inicia em idade tardia, a coreia tende a ser mais proeminente que a lentidão ou a rigidez. Nestes casos, é provável que seja mais difícil estabelecer uma história familiar, porque os pais do doente podem já ter falecido, talvez antes de eles próprios terem apresentado sinais da doença. Os doentes com DH não falecem como resultado direto da doença, mas sim devido a problemas médicos resultantes da condição debilitada do corpo. Estes incluem a pneumonia (que representa um terço de todas as mortes em doentes com DH), engasgamento, falência cardíaca, traumatismo craniano resultante de quedas, e deficiências nutricionais. O risco de suicídio é significativamente elevado, estando associado a mais de 7% das mortes.Quanto comum é a DH?
Quais são os sintomas da DH?
Os sintomas psiquiátricos mais comuns da DH são a apatia, ansiedade, depressão, irritabilidade, explosões de raiva, impulsividade, comportamentos obsessivo-compulsivos, perturbações do sono e isolamento social. Reporta-se menos frequentemente mania e esquizofrenia – incluindo delírios (ideias falsas) e alucinações (ver, ouvir ou sentir coisas que não existem na realidade). Os indivíduos afetados podem experienciar ideação suicida, especialmente nas primeiras fases da doença. A maior parte dos doentes com DH e os seus cuidadores sentem os sintomas comportamentais com maior ansiedade do que as alterações motoras ou cognitivas causadas pela doença.
A DH é caracterizada pela diminuição gradual da compreensão, raciocínio, julgamento e memória. Os sintomas cognitivos incluem a lentificação do pensamento e a dificuldade de concentração, organização, planeamento, tomada de decisão e resposta a questões, bem como défices na memória de curto prazo e na capacidade de resolução de problemas e alteração na capacidade de adquirir e compreender nova informação.
Existe um conjunto de outras alterações que podem ocorrer durante a DH, incluindo a perda de apetite, de peso, de auto-estima e de líbido, assim como incontinência urinária e fecal.Quando aparecem os sintomas da DH?
O que determina a idade de início dos sintomas?
Quais são os diferentes estadios de progressão da DH?
Os sintomas da DH juvenil diferem dos sintomas dos adultos?
Quais são os sintomas quando a DH se inicia na idade adulta tardia?
Causas de morte
Diagnóstico & Tratamento
Como é diagnostica a DH?
A DH é diagnosticada por uma combinação de avaliações clínicas e de um teste genético. O diagnóstico clínico é baseado na história clínica e familiar da pessoa, bem como em observações padrão que utilizam escalas clínicas para avaliar a frequência e a gravidade dos sintomas da DH. Os resultados do diagnóstico clínico são geralmente confirmados pelo estudo genético da expansão do gene HTT (conhecido como teste genético diagnóstico ou de confirmação). Se a pessoa não apresentar sintomas, mas estiver em risco de ser portador da doença, um teste genético em pessoa assintomática (conhecido por teste genético preditivo) determina se a pessoa é ou não portadora da expansão.
Os instrumentos de avaliação clínica utilizados para diagnosticar a DH e medir a forma de apresentação da doença não são os mesmos em todos os centros clínicos ou em todos os países. Contudo, o instrumento mais frequentemente utilizado é a Escala Unificada da doença de Huntington (Unified Huntington’s Disease Rating Scale (UHDRS)), que se encontra dividida nas subsecções motora, comportamental, cognitiva e de funcional. Também a Escala de Avaliação de Problemas Comportamentais para a doença de Huntington (Problem Behaviours Assessment for Huntington’s Disease (PBA)) é frequentemente utilizada para medir a gravidade e a frequência das alterações comportamentais (como o humor depressivo, apatia ou a irritabilidade) enquanto que alguns testes como a Avaliação Breve do Estado Mental (Mini-Mental State Examination (MMSE)) e a Escala de Avaliação de Demência Mattis são usados como complemento da sub-categoria da UHDRS que avalia as alterações cognitivas.Que ferramentas de avaliação são utilizadas no diagnóstico da DH?
Viver com a consciência de estar em risco de vir a ter DH pode ser um fator de grande ansiedade. Pode sentir que preferia saber com certeza se é portadora da expansão. Nesse caso, são altamente recomendados aconselhamento genético e apoio psicológico, visto permitirem-lhe explorar todas as opções e discutir as suas preocupações. Em geral, o teste preditivo não é aconselhado a menores de 18 anos – idade na qual se espera que a pessoa tenha maturidade para lidar com o conhecimento de ser portador da expansão. Em casos excecionais, contudo, pode considerar-se razoável um teste genético confirmatório a crianças – se apresentarem sinais de HD juvenil, por exemplo – ou a mulheres jovens com menos de 18 anos se estiverem grávidas. Se decidir fazer o teste, será colhida uma amostra de sangue de uma veia no braço e o seu ADN extraído desta amostra no laboratório. Dependendo do serviço em cada região, o resultado está pronto entre duas a oito semanas. As recomendações para o procedimento para testes genéticos preditivos foram atualizadas pelo Grupo de Trabalho em Testes Genéticos e Aconselhamento da Rede Europeia de doença de Huntington (EHDN), em 2012.Qual é o procedimentos para realizar um teste genético preditivo?
O teste genético determina o número de repetições CAG no gene HTT. O teste pode revelar se uma pessoa é, ou não, portador da expansão, mas não consegue determinar quando a doença irá ter início, com que velocidade irá progredir ou que sintomas essa pessoa irá desenvolver. O teste genético para a DH é considerado quase 100% preciso. Os resultados da análise do ADN são habitualmente duplamente verificados utilizando duas amostras de sangue separadas. Além disso, o sangue de um dos pais da pessoa afetada (ou, se não estiver disponível, de outro membro da família) também pode ser testado para fornecer confirmação do diagnóstico original.O que detecta o teste genético?
Sim. Isto pode ser conseguido usando um procedimento de diagnóstico moderno chamado diagnóstico genético de pré-implantação (PGD) – também conhecido como triagem embrionária – que é usado em conjunto com fertilização in vitro (FIV) e envolve a triagem de embriões antes da implantação no útero. A técnica garante que apenas os embriões que herdam cópias normais do gene são implantados. Assim, mesmo que um deles carregue a mutação da DH, o PGD permite que um casal conceba uma criança que não carrega o gene HTT mutante, independentemente de o portador ser o homem ou a mulher. No entanto, PGD não é permitido em alguns países sob leis de proteção embrionária, e também é importante notar que as hipóteses de uma gravidez chegar a termo após PGD / IVF são menores do que no caso da conceção “natural”. Em alguns países, é possível testar os fetos não nascidos que foram concebidos naturalmente e escolher o aborto quando o estado genético do feto é conhecido.É possível uma pessoa portadora da DH assegurar-se de que não
passará a doença para os seus filhos?
O diagnóstico pré-natal (antes do nascimento) só está disponível quando aqueles que o solicitem podem mostrar que seu caso cumpre certos critérios médicos e legais, que são específicos a cada país. Existem dois procedimentos padrão para o diagnóstico pré-natal. O primeiro é a amniocentese (também chamada de teste de líquido amniótico), na qual o líquido amniótico que contém células fetais é coletado através de uma agulha inserida através da parede abdominal da mãe, geralmente após a 14a semana de gravidez. O segundo é a amostra das vilosidades coriónicas, que envolve a colheita de uma amostra das velosidades corionicas (tecido placentário) e pode ser realizada mais precocemente – entre as 10a e 13a semanas de gravidez. É, no entanto, mais arriscado para o feto.Posso testar o meu filho antes de ele nascer?
Não existem actualmente terapêuticas que possam tratar de forma eficaz as causas subjacentes da DH. No entanto, a investigação básica e clínica realizada nos últimos anos aumentou de forma drástica o conhecimento de doença e muitos estudos em curso investigam sua patogenese, com o objetivo de identificar fármacos que adiem o início da doença ou retardem a sua progressão. Existem tratamentos já disponíveis que aliviam certos sintomas da doença (tratamentos sintomáticos), melhorando assim a qualidade de vida dos doentes. Estes são divididos em tratamentos farmacológicos (medicamentos) e não farmacológicos (não utilizando medicamentos).Existem tratamentos para a DH?
A coreia, a bradicinesia, a irritabilidade, a apatia, a depressão, a ansiedade e os distúrbios do sono foram todos reportados como sendo os sintomas mais angustiantes da DH. Existem várias opções para tratar esses sintomas utilizando medicação. No entanto, muitos medicamentos podem causar efeitos secundários e alguns podem inclusivamente contrariar os efeitos terapêuticos de outros. Além disso, a mesma medicação pode ter efeitos diferentes em diferentes indivíduos. O tratamento deve ser personalizado por um especialista experiente em DH, de acordo com os sintomas do doente e sua resposta à medicação em questão.Quais são as opções farmacológicas para o tratamento dos sintomas
da DH?
Os tratamentos não-farmacológicos (como a psicoterapia e terapias cognitivas, fisioterapia e terapias da fala, respiratória e ocupacional) podem melhorar os sintomas psicológicos e físicos da DH. Foram, por exemplo, reportadas melhorias no humor, controlo motor, fala, equilíbrio, deglutição e na marcha após a realização destas terapias. Sabe-se que o exercício físico melhora a saúde física e mental, aumentando o bem-estar geral e o mesmo tem demonstrado aliviar os sintomas da depressão. Há evidência crescente de que poderá, também, ajudar a diminuir a progressão das alterações do movimento na DH. Por exemplo, alguns programas de fisioterapia demonstraram ter benefícios em termos de sintomas motores, marcha e equilíbrio. O Grupo de Trabalho de Fisioterapia da EHDN publicou um documento de orientação para fisioterapeutas que trabalhem com doentes com DH.Como podem ajudar os tratamentos não-farmacológicos?
Os benefícios de uma dieta rica em vitaminas, coenzimas e outros compostos tem sido objeto de muita discussão, mas continuam por ser comprovados clinicamente. No entanto, como a perda de peso é um problema para alguns doentes com DH, principalmente nos estadios mais avançados da doença, é importante garantir uma dieta saudável ao longo de todo o curso da doença. Nos estadios avançados, uma dieta com alto teor calórico pode tornar-se necessária. Pode ser útil a referenciação a um nutricionista.Uma dieta especial pode aliviar os sintomas da DH?
Hereditariedade & o que causa a DH
A DH é causada por uma mudança (uma expansão) no gene (HTT) que codifica uma proteína chamada huntingtina. Como resultado desta expansão, o gene é traduzido numa forma alterada da proteína, algo que leva ao mau funcionamento e morte de células nervosas (neurónios) em áreas específicas do cérebro. Os mecanismos exactos da doença são multifacetados e altamente complexos, de acordo com as múltiplas funções da proteína huntingtina. Os investigadores têm trabalhado para conseguir ter uma melhor compreensão dos mecanismos causadores da doença de forma a desenvolverem terapias modificadoras da doença.O que causa a DH?
Os cientistas identificaram em 1993 a mutação que causa a DH. O gene HTT está localizado no cromossoma 4 e codifica uma proteína chamada huntingtina. O gene contém uma sequência de três nucleótidos (as unidades base do ADN), citosina-adenina-guanina (CAG), que é repetida várias vezes. Essa chamada repetição de trinucleótidos pode variar em comprimento. Se uma pessoa tiver 40 ou mais de 40 repetições CAG numa cópia do gene HTT, desenvolverá DH dentro de um tempo de vida normal – ou seja, na idade adulta média. Uma vez que a expansão que causa a DH está presente em todas as células do corpo desde a concepção, e pode ser transmitida para gerações subsequentes, a DH é uma doença hereditária.Qual é a causa subjacente da DH?
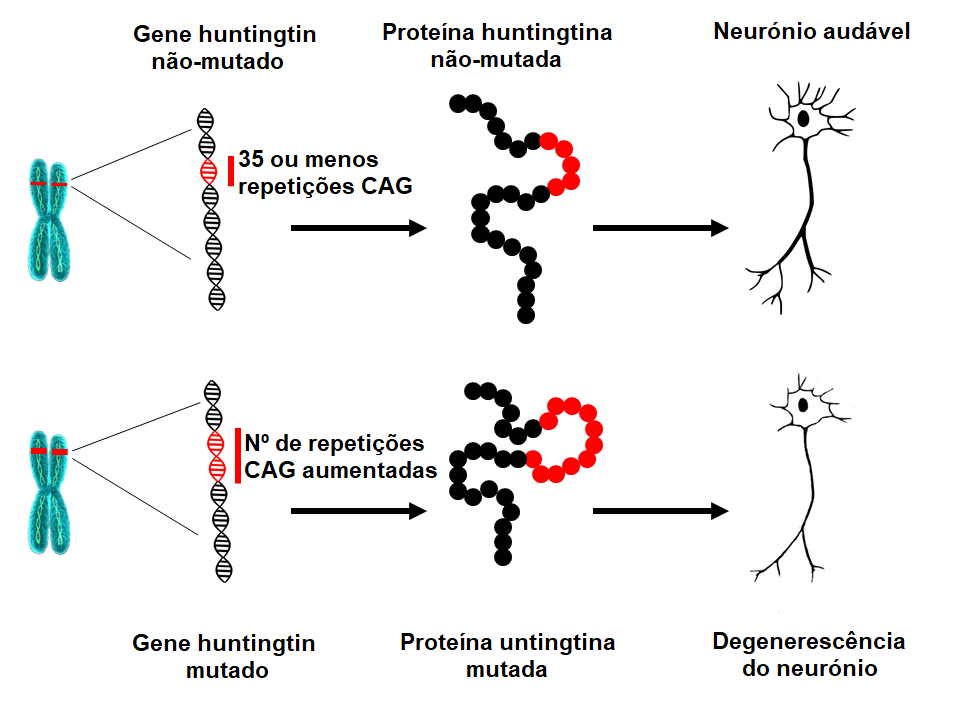
À medida que o número de repetições CAG aumenta, essa secção particular de DNA torna-se mais instável. Isto significa que o número de repetições CAG nesta secção pode aumentar ou diminuir quando é transmitida à geração seguinte. Desde que o número de repetições CAG do gene HTT esteja abaixo de 27, a secção é estável. Se o número de repetições está entre 27 e 35 (o chamado comprimento intermédio de repetições), esse individuo não irá desenvolver a doença e a secção é considerada normal. Contudo, um número de repetições de CAG de 27 ou mais é instável e está sujeita a aumentar quando for transmitida para a próxima geração, o que quer dizer que essas crianças têm um risco de desenvolver doença de Huntington. Indivíduos com um número de repetições CAG entre 36 e 39 podem desenvolver DH, mas só muito tardiamente na vida, se alguma vez desenvolverem. Isto é conhecido como o comprimento de repetições de baixa penetrância. Quando o número de repetições de CAG é superior a 39, a pessoa vai desenvolver DH dentro de um tempo de sobrevida normal – mais frequentemente em idade adulta média. Em casos raros, a expansão CAG pode ser excecionalmente longa, levando ao início da doença na adolescência ou na infância (DH Juvenil). Os doentes que desenvolvem a doença antes dos 10 anos têm frequentemente mais de 80 repetições CAG.O que significa o comprimento das repetições CAG do gene HTT?
Número de repetições CAG Provoca doença? Consequências para a descendência Nome
Abaixo de 27 Não Nenhuma Número de repetições normal
27-35 Não Um número de repetições maior ou igual a 27 pode ser instável e pode aumentar quando passado à descendência Número de repetições intermédio
36-39 Talvez Sim, cada um dos filhos tem 50% de probabilidade de herdar o gene expandido Número de repetições de penetrância reduzida
40 ou acima de 40 Sim Sim, cada um dos filhos tem 50% de probabilidade de herdar o gene expandido Número de repetições de penetrância completa
Os genes estão nos nossos cromossomas, dentro de todas as células do nosso corpo. Um gene é uma porção do ADN que contém um código para uma determinada proteína; o DNA é transcrito em RNA mensageiro (mRNA), que é de seguida traduzido numa proteína. Todos herdamos habitualmente duas cópias de cada gene – uma da mãe, outra do pai. Na DH, o gene importante é o gene HTT, que codifica a proteína huntingtina. Quando uma criança herda uma versão expandida do gene HTT também vai desenvolver DH. O pai ou a mãe pode já ter a doença, ou a mesma pode desenvolver-se numa idade mais tardia.O que é um gene?
As proteínas são moléculas grandes formadas por blocos de construção denominados aminoácidos. A sequência exata de aminoácidos numa proteína específica é determinada pela sequência de ADN do gene correspondente. Os genes funcionam, portanto, como planos – conjuntos de instruções para células que lhes dizem como criar proteínas específicas. O gene HTT contém instruções sobre como construir a proteína huntingtina. As proteínas são as moléculas que fazem o trabalho dentro das células – realizam uma grande quantidade de processos essenciais, como reações enzimáticas ou suporte estrutural. Se uma proteína funciona anormalmente ou está em falta devido a uma expansão no gene que a codifica, então ela pode afetar a célula e, em última instância, o organismo inteiro, causando, por vezes, doenças.O que é uma proteína?
A proteína huntingtina é uma proteína muito grande que é produzida ou “expressa” em variados graus em todas as células do corpo humano; os níveis mais altos são encontrados no cérebro. A huntingtina parece ser uma proteína muito importante porque a sua ausência é letal para embriões de ratinho. Numa das extremidades da proteína huntingtina há uma porção de repetições de um aminoácido particular chamado glutamina. Esta característica distintiva, conhecida como a repetição de poliglutamina, consiste normalmente em até 35 unidades de glutamina. Nas pessoas portadoras da expansão da DH, no entanto, contém pelo menos 36 repetições e é essa expansão de poliglutamina que resulta numa proteína que funciona mal.A proteína huntingtina
A HD é uma doença hereditária dominante. Isto significa que uma pessoa nascida com uma cópia do gene HTT mutante desenvolverá a DH mesmo que também seja portador de uma cópia normal do gene. Um portador da expansão da DH, seja sintomático ou não, pode transmitir ou uma cópia normal ou uma cópia com a mutação do gene, com uma probabilidade de 50% de cada um dos casos (desde que a pessoa só seja portadora da expansão numa das duas cópias do gene HTT). Existem técnicas médicas que podem garantir que um indivíduo afetado apenas transmita o gene HTT normal aos seus filhos. Por outro lado, uma pessoa que não herdou o gene HTT mutado não desenvolverá a doença, e seus filhos também não estarão em risco. A expansão da DH não pode saltar uma geração. No entanto, pode acontecer que um portador da expansão morra antes de desenvolver sintomas e que seus filhos não percebam que estão em risco de desenvolver a doença.Como
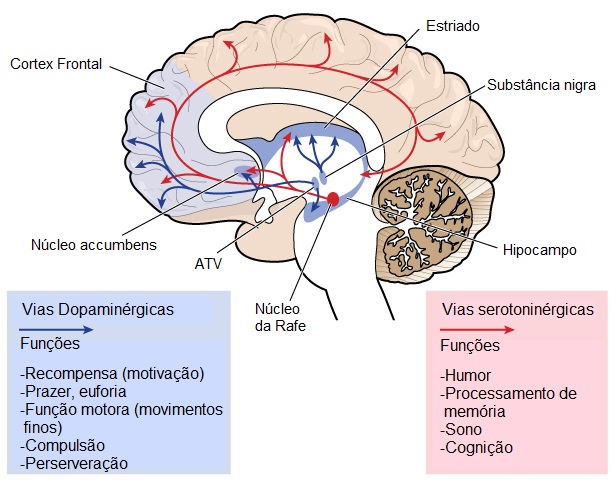
Certas funções do cérebro, como a capacidade de se movimentar, pensar e falar deterioram-se gradualmente na DH, à medida que células nervosas cruciais se danificam e morrem. A parte do cérebro mais afetada pela HD é o estriado, que faz parte dos gânglios basais e que se localiza na região central profunda do cérebro. O estriado está principalmente envolvido no planeamento e controlo dos movimentos, mas também em muitos outros processos, incluindo cognição e emoções. À medida que a DH progride, afeta o córtex (a parte mais exterior e enrugada do cérebro), contribuindo para a deterioração cognitiva. Em geral, a DH causa atrofia de todo o cérebro ao longo do tempo, prejudicando a capacidade funcional geral do indivíduo.Áreas cerebrais envolvidas na DH
A DH na vida diária
Ter um resultado positivo para a expansão da DH pode afetar diferentes aspetos da vida de uma pessoa, incluindo a decisão de ter ou não filhos, planear o futuro, repensar prioridades, negociar habitação apropriada e informar outros familiares de que também eles podem estar em risco de desenvolver a doença. Os Jovens adultos, em particular, podem precisar de ter em consideração as implicações de um resultado genético positivo na sua educação, formação e emprego. À medida que a doença progride, a capacidade da pessoa para viver de forma independente é gradualmente afetada. Trabalho, vida social e atividades de vida diária, quotidiana, tornam-se problemáticos e os doentes tornam-se progressivamente dependentes da ajuda tanto de familiares, como de profissionais de saúde e de ação social. Podem-se contactar, em qualquer altura, grupos ou associações de doentes e centros clínicos locais que providenciarão suporte.De que forma a DH afecta a vida diária?
Estratégias eficientes para lidar com a DH têm de ser pensadas personalizadamente e consoante a pessoa afetada, o estadio da doença em que se encontra e o contexto familiar. A DH progride muito lentamente, de forma que, geralmente, há tempo para adaptação às alterações de induz. Para os cuidadores e pessoas próximas, uma melhor compreensão das alterações do comportamento e cognitivas, do pensamento, associadas à doença pode ajudar a desenvolver estratégias que integrem estas mudanças na rotina e permitam a manutenção de um bom relacionamento com a pessoa doente. Especialistas em DH e grupos ou associações de doentes podem dar informações e conselhos úteis.Existem estratégias para lidar melhor com a DH?
Como contacto a EHDN?
A marcação pode ser feita através do encaminhamento pelo seu médico de família ou contactando o coordenador de linguagem da EHDN mais próximo de si.Como faço uma marcação com um especialista?
Associações ou grupos de apoio de doentes do seu país podem dar um aconselhamento independente fora do contexto de uma clínica.Existe alguma forma de falar com um especialista sem ter de me dirigir
a uma clínica?
A EHDN tem um papel chave no estudo clínico mundial Enroll-HD. Trata-se de um estudo observacional que não envolve qualquer intervenção. Isto significa que não testa terapias experimentais. Os participantes realizam avaliações clínicas durante as suas visitas anuais e podem ser elegíveis a participar em ensaios clínicos de terapias sintomáticas ou modificadoras de doença quando e à medida que estas novas terapias estejam disponíveis. O Enroll-HD é acessível através de muitos centros de estudo da DH distribuídos todo o mundo. Para saber se existe um centro perto de si, por favor procure aqui ou contacte o seu coordenador de linguagem da EHDN, que lhe pode dar informações sobre as atividades de investigação na sua região. As associações de doentes ou grupos de apoio da sua região podem providenciar informação generalizada acerca da participação em investigações. Para informações adicionais sobre investigação na DH, por favor procure aqui ou visite a página HDBuzz na internet, que apresenta novidades escritas por investigadores da DH em linguagem acessível ao público em geral e traduzida para quase todas as línguas.Como me posso envolver na investigação em DH?
Sim, existem várias associações ou grupos de apoio a doentes que dão apoio a pessoas e famílias afetadas pela DH. Podem ser contactados através do seu médico de família ou do seu especialista em DH, ou pode contacta-los diretamente. A Associação Europeia da Doença de Huntington (EHA) tem uma lista de associações e de grupos de apoio a doentes que lhe pode ser útil.Existem grupos de apoio especializados em DH?
Para mais informações, contacte o seu coordenador de linguagem da EHDN, a associação de doentes ou grupo de apoio a doentes da sua área.