HS historik
HS är uppkallat efter George Huntington, en amerikansk läkare som beskrev sjukdomen år 1872. Hans beskrivning baserades på observationer av HS-drabbade familjer från byn East Hampton på Long Island i New York (USA), där Dr. Huntingtons bodde och arbetade. HS var tidigare känt som Huntingtons Chorea och Saint Vitus’s Dans eller danssjukan.
Symtom och sjukdomsutveckling
Huntingtons sjukdom (HS) är en sällsynt sjukdom som finns hos mellan 5 till 10 per 100000 invånare bland den europeiska befolkningen. En snarlik prevalens finns i länder vars befolkning har ett europeiskt ursprung, exempelvis USA. HS är mindre vanligt i asiatiska och afrikanska länder med en prevalens på 1 per 100 000 invånare. Män och kvinnor löper lika stor risk att ärva HS genexpansionen och att utveckla sjukdomen. HS karaktäriseras av en kombination av motoriska (rörelser), psykiatriska (ex. humör) och kognitiva (ex. förståelse) störningar, men andra symtom kan också förekomma. Sjukdomsbilden vid HS varierar från individ till individ och även mellan familjemedlemmarmed med olika grad av funktionsnedsättning, ålder för sjukdomsdebuten och sjukdomsprogression. En individ med HS kan ha en tydlig rörelsepåverkan med en mild psykiatrisk och kognitiv påverkan medan en annan individ med HS kan drabbas av depression och ångest flera år innan motoriska störningar. Sjukdomsdebuten beskrivs ofta som ”smygande” eftersom det ofta är svårt att fastställa ett exakt datum för sjukdomsdebuten. De flesta individer som bär på HS genexpansionen utvecklar symtom i mitten av vuxen livet – det vill säga mellan 35 och 55 års ålder. Cirka 10% utvecklar symtom innan de fyllt 20 (de har juvenil HS) och ytterligare 10% utvecklar symtom efter 55 års ålder. I allmänhet utvecklas HS gradvis och individer kan därför gå odiagnostiserade under många år. I genomsnitt så är sjukdomstiden mellan 15 till 20 år från diagnos men detta varierar mellan olika individer och kan bero på kvaliteten på vården som patienten erhåller. Faktorerna som avgör sjukdomsdebuten är komplexa och föremål för pågående forskning. När man tittar på stora grupper av patienter med HS hittar forskare ett samband mellan antalet trinukleotid repetitioner och ålder vid sjukdomsdebuten (figur nedan). Detta innebär i allmänhet att ju högre antal CAG repetitioner, desto tidigare sjukdomsdebut (se figur nedan). Däremot kan variationerna i åldern för sjukdomsdebut variera med uppemot 30 år för ett visst antal CAG repetitioner, vilket visas i figuren nedan. Detta beror förmodligen på andra gener utöver huntingtingenen (HTT) (så kallade genetiska modifierare) och miljöfaktorer såsom livsstil och kost. Sammantaget är det därför mycket svårt att exakt förutsäga ålder för sjukdomsdebut för en individ som bär på anlaget. Enligt en klassifikation som utvecklats av neurologen och HS specialisten Ira Shoulson vid Georgetown University i USA, kan utvecklingen av HS indelas i fem faser: När HS startar tidigt i livet (före 20 års ålder) är ofrivilliga rörelser (chorea) mindre framträdande än långsamma rörelser (bradykinesi) och stelhet (dystoni). Tidiga symtom vid juvenil HS inkluderar tydliga beteendeförändringar, svårigheter med inlärning och tal och försämrade skolprestationer. Epileptiska anfall har rapporterats och är vanligare hos unga patienter. I allmänhet fortskrider den juvenila formen av sjukdomen snabbare än vuxenformen. När HS startar sent i livet tenderar chorea att vara ett mer framträdande symtom än långsamhet eller stelhet. I fall med sen sjukdomsdebut kan det ibland vara svårare att fastställa en familjehistoria eftersom individens föräldrar kanske redan har dött och kanske även innan de själva visade tecken på sjukdomen. Personer med HS dör inte som en direkt följd av sjukdomen utan snarare från medicinska problem som uppstår till följd av kroppens försvagade tillstånd. Orsakerna inkluderar lunginflammation (som står för en tredjedel av alla dödsfall i HS-patienter), kvävning, hjärtsvikt, skallskada till följd av fall och näringsbrist. Självmordsrisken ökar och svarar för upp till 7% av alla dödsfall bland HS patienter.Hur vanligt är Huntingtons sjukdom?
Vilka är symtomen vid Huntingtons sjukdom?
De vanligaste psykiatriska symtomen i HS är apati, ångest, depression, irritabilitet, aggressionsutbrott, impulsivitet, tvångsmässiga beteenden, sömnstörningar och social tillbakadragenhet. Mer sällsynta men förekommande är mani och schizofreni – inklusive vanföreställningar (felaktiga uppfattningar) och hallucinationer (att se, höra eller känna saker som inte riktigt finns). Drabbade individer kan uppleva självmordstankar, särskilt i tidiga stadier av sjukdomen. De flesta HS patienter och deras anhörigvårdare upplever att beteendeförändringar är mer påfrestande än de motoriska eller kognitiva funktionshinder orsakade av sjukdomen.
HS kännetecknas av en gradvis försämring av fattningsförmåga, resonemang, omdöme och minne. Kognitiva symptom inkluderar långsammare tänkande och svårigheter med koncentration, beslutsfattande och att organisera, planera och svara på frågor. Korttidsminnet och problemlösningsförmågan försämras och sjukdomen leder till en nedsatt förmåga att uppfatta och förstå ny information.
Det finns ett antal andra förändringar som kan uppstå under sjukdomsförloppet, inklusive förlust av aptit, viktminskning, förlust av självkänsla, förlust av sexlust samt urinvägs- och fekal inkontinens.När uppkommer HS symtomen?
Vad avgör sjukdomsdebuten?
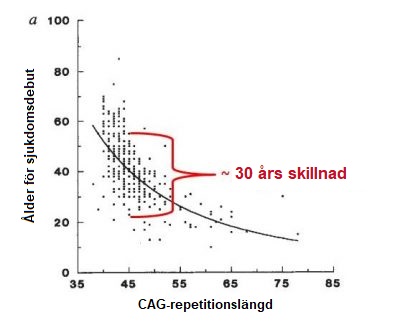
Nature Genetics 4, 398–403 (1993).Vilka är de olika faserna i sjukdomsutvecklingen?
Skiljer sig symtomen mellan den juvenila och vuxenformen av HS?
Vilka är symtomen vid sen debut av Huntingtons sjukdom?
Dödsorsaker
Diagnos & behandling
HS diagnostiseras genom en kombination av kliniska bedömningar och ett genetiskt test. Kliniska diagnosen baseras på en persons medicinska historia och familjehistoria samt genom standard undersökningar med kliniska skattningsskalor som används för att bedöma frekvensen och svårighetsgraden av HS symtomen. Resultaten av den kliniska diagnosen bekräftas vanligtvis av genetisk testning av anlaget för HS (även kallat diagnostisk genetisk testning). Om en person inte visar några symtom, men är i riskzonen för att utveckla sjukdomen, kan asymtomatisk genetisk testning (prediktiv genetisk testning) avgöra om de bär anlaget för HS.Hur diagnostiseras HS?
De kliniska bedömningsverktyg som används för att diagnostisera och mäta olika aspekter av HS kan skilja sig mellan olika kliniker och länder. Det mest använda verktyget heter Unified Huntingtons sjukdom Rating Scale (UHDRS) och delas in i motoriska, beteendemässiga, kognitiva och funktionella utvärderingar. Dessutom används Problem Behaviours Assessment for Huntington’s Disease (PBA) ofta för att bedöma svårighetsgraden och frekvensen av beteendeavvikelser (såsom nedstämdhet, apati och irritabilitet), medan en mängd tester såsom Mini-Mental State Examination (MMSE) och Mattis Dementia Rating scale används för att komplettera delen i UHDRS som bedömer kognitiva funktionsnedsättningar.Vilka kliniska tester används för att diagnostisera HS?
Att leva med vetskapen om att du är i riskzonen för HS kan vara mycket oroande. Du kanske känner att du hellre vill veta om du bär på anlaget för HS. I så fall rekommenderas genetisk rådgivning och psykologiskt stöd, eftersom dessa forum tillåter dig att utforska dina alternativ och diskutera din oro. Prediktiv genetisk testning rekommenderas i allmänhet inte för dem som är yngre än 18 ålder, vilket anses vara den ålder då en person är mogen att hantera vetskapen om de bär på anlaget för HS. I undantagsfall kan testet utföras på barn om de visar tecken på juvenil HS eller kvinnor, som är yngre än 18 år, om de är gravida. Om du väljer att testa dig tar man ett venöst blodprov från din arm och extraherar ditt DNA från blodprovet i ett laboratorium. Beroende på den lokala servicen får man vanligtvis resultatet inom 2-8 veckor. Riktlinjer för prediktiv genetisk testning uppdaterades 2012 av EHDN Genetic Testing and Counselling Working Group.Vad är proceduren för prediktiv genetisk testning?
Det genetiska testet bestämmer antalet CAG repetitioner i huntingtingenen. Testet kan avslöja om du bär på anlaget för HS, men det kan inte fastställa när sjukdomen kommer att bryta ut, hur snabbt den kommer att utvecklas eller vilka symtom du kommer att utveckla. Det genetiska testet för HS anses vara nära 100% korrekt. Resultatet av DNA-analysen bekräftas vanligtvis genom att analysera två separata blodprov. Dessutom kan ett blodprov från en förälder till den drabbade personen (eller från annan familjemedlem) analyseras för att bekräfta den ursprungliga diagnosen.Vad bestämmer det genetiska testet?
Ja. Detta kan uppnås med hjälp av en modern diagnostisk procedur som kallas preimplantatorisk genetisk diagnostik (PGD), som används i kombination med in vitro-fertilisering (IVF) och som innebär screening av befruktade ägg innan de återförs till kvinnans livmoder. Tekniken garanterar att endast befruktade ägg med normala kopior av genen återförs. PGD gör det möjligt för ett par att få ett barn som inte bär på HS anlaget oavsett om det är mannen eller kvinnan som bär på anlaget. PGD är inte tillåtet i vissa länder och det är också viktigt att notera att chansen för graviditet är lägre vid PGD/IVF jämfört med ”naturlig” befruktningen. I vissa länder är det möjligt att testa ofödda foster som har befruktats på naturlig väg med möjlighet för abort när fostrets genetiska status är känd.Är det möjligt för en person som bär på anlaget för HS att säkerställta
att inte föra anlaget vidare till sitt barn?
Fosterdiagnostik finns oftast tillgängligt och följer nationella riktlinjer. Det finns olika metoder som används vid fosterdiagnostik. En metod är fostervattenprov där fostervattnet som innehåller fostrets celler samlas in via en tunn nål som förs in genom moderns bukvägg. Detta görs vanligtvis efter den 14:e graviditetsveckan. En annan metod är moderkaksprov, som innebär insamling av vävnad från moderkakan. Detta test kan utföras tidigare – mellan den 10:e och 13:e veckorna av graviditeten. Det finns däremot en ökad risk för missfall.Kan jag testa mitt ofödda barn?
Det finns för närvarande inga behandlingar som effektivt kan behandla de underliggande orsakerna till HS. Grundforskning och klinisk forskning har emellertid under de senaste åren ökat kunskapen om HS dramatiskt. Många studier som pågår nu undersöker sjukdomsmekanismerna i syfte att identifiera läkemedel som kan senarelägga sjukdomsdebuten eller bromsa utvecklingen av sjukdomen. Det finns redan behandlingar som lindrar vissa av symtomen (sk symtomatiska behandlingar) och som därmed förbättrar patienternas livskvalitet. Dessa är indelade i farmakologiska (läkemedel) och icke-farmakologiska (utan läkemedel) behandlingar.Finns det behandlingar för HS?
Chorea, bradykinesi, irritabilitet, apati, depression, ångest och sömnstörningar har rapporterats vara de mest plågsamma symptomen i HS. Det finns flera alternativ för att hantera dessa symtom med hjälp av läkemedel. Däremot kan många läkemedel orsaka biverkningar och några kan motverka effekterna av andra läkemedel. Dessutom kan samma läkemedel ha olika effekter i olika individer. Behandlingen måste individanpassas av en erfaren specialist på HS som tar både patientens symtom och hur patienten svarar på ett läkemedel i beaktande.Vilken farmakologisk behandling finns för att lindra HS symtom?
Icke-farmakologiska behandlingar (såsom psykoterapi och kognitiva, fysio- tal-, respiratoriska och arbetsterapier) kan förbättra de psykiska och fysiska symptom vid HS. Exempelvis har man observerat förbättringar i humör, motorik, tal, balans, sväljförmåga och gång som följd av dessa behandlingar. Det är väl känt att fysisk träning förbättrar både fysisk och psykisk hälsa och det allmänna välbefinnandet. Motion har visat sig lindra depression och det finns även bevis för att motion kan hjälpa till att bromsa utvecklingen av motoriska symtom i HS. Till exempel har vissa sjukgymnastik program visat sig resultera i gynnsamma effekter på motoriska symtom, gång och balans. EHDN Physiotherapy Working Group har publicerat en vägledning för sjukgymnaster som arbetar med HS-patienter.Hur hjälper icke-farmakologiska behandlingar?
Fördelarna med en kost rik på vitaminer, coenzymer och andra substanser har varit föremål för mycket diskussion, men det återstår att bevisa effekterna av dessa kliniskt. Då viktminskning är ett problem för vissa HS patienter, särskilt i de senare stadierna av sjukdomen, så är det viktigt att säkerställa en hälsosam kost under hela sjukdomen. I senare skeden av sjukdomen kan en kaloririk kost bli nödvändig. Remiss till en dietist kan vara till hjälp.Kan vissa dieter förbättra symtomen i HS?
Ärftlighet & Vad orsakar HS?
HS orsakas av en förändring (en expansion) i den gen (HTT) som kodar för ett protein som kallas huntingtin. Till följd av denna expansion översätts genen till ett protein med förändrad form vilket förstör funktionen och leder till död av nervceller (neuroner) i specifika områden i hjärnan. De exakta mekanismerna av sjukdomen är mångfacetterade och komplexa och följer huntingtinproteinets många funktioner. Forskare arbetar på att få en bättre förståelse av de underliggande sjukdomsmekanismerna för att kunna utveckla sjukdomsmodifierande terapier.Vad orsakar HS?
Forskare identifierade mutationen som orsakar HS år 1993. HTT genen finns på kromosom 4 och kodar för ett protein som kallas huntingtin. Genen innehåller en sekvens av tre nukleotider (de grundläggande enheterna av DNA), cytosin-adenin-guanin (CAG), som upprepas flera gånger. Denna så kallade trinukleotid repetition kan variera i längd. Om en person har 40 CAG-repetitioner eller mer i en kopia av genen HTT kommer han/hon att utveckla HS inom en normal livslängd – det vill säga i mitten av vuxenlivet. HS är en ärftlig sjukdom eftersom anlaget som orsakar HS finns i alla celler i kroppen från befruktningen och kan nedärvas vidare till kommande generationer.Vad är orsaken till HS?
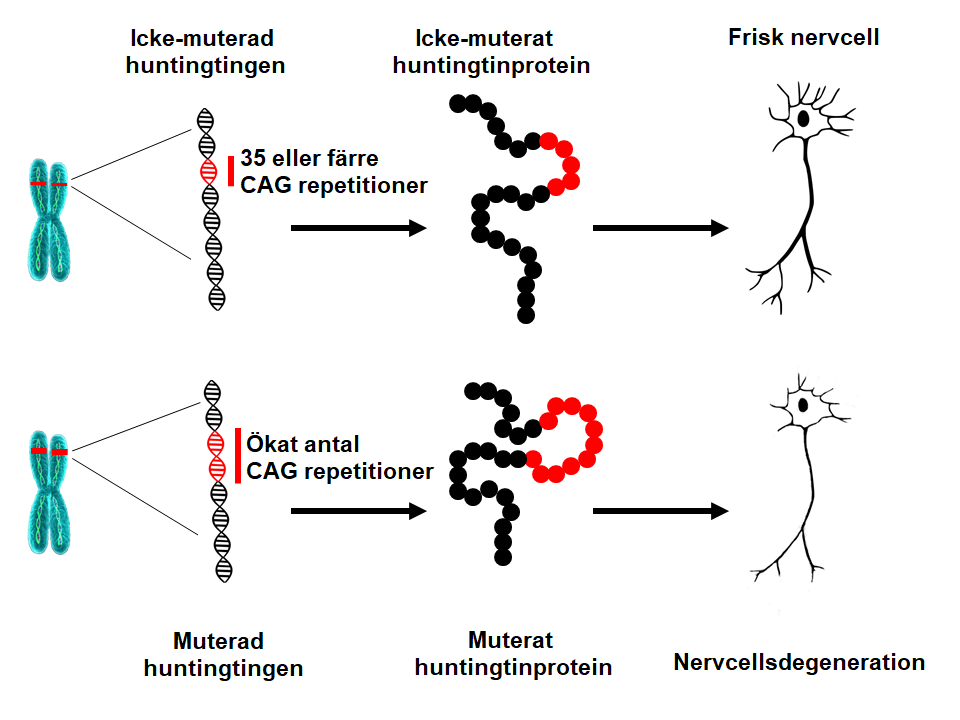
När antalet CAG repetitioner ökar blir den delen av DNA:t mer instabilt. Detta innebär att antalet repetitioner i den här regionen kan öka eller minska när det förs vidare till nästa generation. Så länge antalet CAG repetitioner i HTT genen är lägre än 27 är regionen stabil. Om antalet repetitioner är mellan 27 och 35 (intermediär repetitionslängd) anses regionen vara normal och individen kommer inte att utveckla HS. Dock kommer en region med 27 eller flera CAG repetitioner att vara mer instabil och det finns därmed en möjlighet för expansion när den förs vidare till nästa generation vilket innebär att dessa barn löper en risk att utveckla HS. Individer med 36-39 CAG repetitioner kan utveckla HS, men endast mycket sent i livet om alls. Intervallet 36-39 kallas för det reducerade penetrans repetitionsintervallet. När antalet CAG-repetitioner är högre än 39 kommer en person utveckla HS inom en normal livslängd – oftast i mitten av vuxenlivet. I sällsynta fall kan CAG expansion vara exceptionellt lång vilket leder till sjukdomsdebut i barndom eller i tonåren (juvenil HS). Patienter som utvecklar sjukdomen innan 10 års ålder har ofta fler än 80 CAG-repetitioner.Vad innebär längden på HTT CAG repetitionerna?
CAG-repetitionslängd
Orsakar sjukdom?
Konsekvenser för avkomma?
Namn
Under 27
Nej
Nej
Nej
27-35
Nej
27 eller flera repetitioner kan vara instabila och kan öka i antal vid nerärvning
Intermediär penetrans
36-39
Kanske
Ja. Avkomma löper 50% risk att ärva anlaget.
Reducerad penetrans
40 eller flera
Ja
Ja. Avkomma löper 50% risk att ärva anlaget.
Fullt penetrant
Gener finns på våra kromosomer i varje cell i vår kropp. En gen är en del av DNA som innehåller koden för ett visst protein. DNA översätts till budbärar-RNA (mRNA), som sedan översätts till proteinet. Oftast ärver alla två kopior av varje gen, en från mamman och en från pappan. I HS är den viktiga genen HTT genen, som kodar för huntingtinproteinet. När ett barn ärver en expanderad version av HTT genen kommer barnet att utveckla HS. Föräldern har själv kanske redan utvecklat sjukdomen eller så kan sjukdomen utvecklas i senare ålder.Vad är en gen?
Proteiner är stora molekyler som består av byggstenar som kallas aminosyror. DNA-sekvensen av en gen bestämmer den exakta ordningsföljden av aminosyror i ett protein. Gener fungerar därför som ritningar – de ger instruktioner till cellerna om hur man bygger specifika proteiner. HTT genen innehåller instruktioner om hur man bygger huntingtinproteinet. Proteiner utgör de molekyler som gör arbetet inuti cellerna – de utför många viktiga processer, som exempelvis enzymreaktioner och strukturellt stöd. Om ett protein fungerar onormalt eller om det saknas på grund av en expansion i genen som kodar för proteinet så kan detta leda till att cellerna, och i slutändan även hela organismen påverkas, vilket kan orsaka sjukdom.Vad är ett protein?
Huntingtinproteinet är ett mycket stort protein som uttrycks i varierande grad i varje cell i människokroppen. De högsta nivåerna finns i hjärnan. Huntingtinproteinet verkar vara ett mycket viktigt protein eftersom avsaknad av proteinet leder till att musembryon dör. I en viss del av huntingtinproteinet finns ett område med flera enheter av aminosyran glutamin. Detta område kännetecknas som en polyglutamin-repetition och består normalt av upp till 35 glutaminenheter. Hos personer som bär på anlaget för HS så innehåller det här området minst 36 repetitioner och det är denna polyglutaminexpansion som resulterar i en felaktig form av proteinet.Huntingtinproteinet
HD är en dominant ärftlig sjukdom. Detta innebär att en person som är född med en kopia av den muterade HTT genen kommer att utveckla HS även om han/hon också bär en normal kopia av genen. En bärare av HS genexpansion, oavsett om hen är symtomatisk eller inte, kan föra vidare den muterade genkopian med 50% risk (förutsatt att han/hon endast bär på genexpansionen på en av de två kopiorna av HTT genen). Det finns medicinska tekniker som kan säkerställa att en drabbad individ endast för vidare den normala genkopian till hans/hennes barn. En person som inte ärvt den muterade genkopian kommer inte att utveckla sjukdomen och hans/hennes barn löper heller ingen risk att insjukna i HS. Anlaget för HS hoppar inte över generationer. Det kan dock hända att en anlagsbärare för HS dör innan hen utvecklar symtom och att hans/hennes barn inte förstår att de löper en risk att utveckla sjukdomen.Hur nedärvs HS?
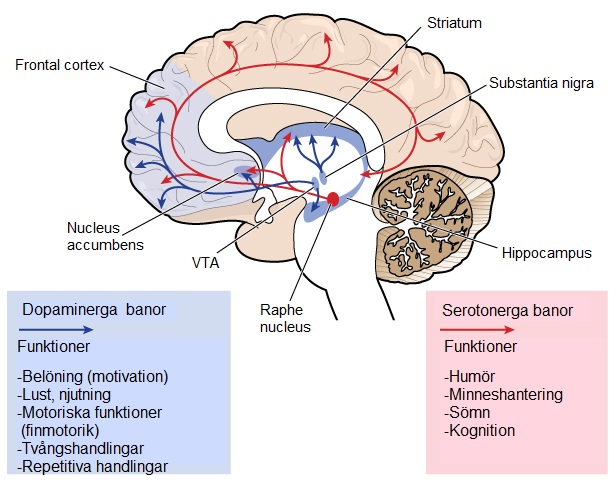
När viktiga nervceller skadas och dör påverkas vissa funktioner i hjärnan med en gradvis försämring av talförmågan och möjligheten att röra sig och att tänka. Striatum är den del av hjärnan som påverkas mest i HS. Striatum är en komponent av de basala ganglierna och ligger djupt i hjärnans centrala region. Striatum är primärt inblandad i planering och styrning av rörelser, men också i många andra processer inklusive kognition och känslor. Allteftersom sjukdomen fortskrider påverkas cortex (den yttersta skrynkliga delen av hjärnan) vilket leder till en kognitiv försämring. HS orsakar med tiden atrofi av hela hjärnan vilket försämrar individens allmänna funktionella kapacitet.Hjärnregioner som påverkas i HS
HS i dagliga livet
Att testa positivt för anlaget för HS kan påverka många olika aspekter av en persons liv inklusive beslut om att skaffa barn eller inte, planering för framtiden, omvärdera prioriteringar, bestämma om lämplig bostad och att informera andra familjemedlemmar om att de kan löpa en risk för att utveckla sjukdomen. Unga vuxna kan särskilt behöva överväga konsekvenserna av ett positivt testresultat för deras utbildning och sysselsättning. När sjukdomen fortskrider påverkar det gradvis en persons förmåga att leva ett självständigt liv. Arbetslivet och de sociala och dagliga aktiviteterna blir problematiska och patienter blir alltmer beroende av hjälp från anhöriga och hälso- och omvårdnadspersonal. Lokala patientstödgrupper och kliniska centra kan kontaktas för stöd.Hur påverkar HS det dagliga livet?
Strategier för att klara HS måste individanpassas och beror på den drabbade personen, sjukdomsfas och familjesituation. HS utvecklas mycket långsamt vilket gör att det i allmänhet finns tid för att anpassa sig till de förändringar som sjukdomen medför. För vårdare och anhöriga kan en bättre förståelse av de beteendemässiga och kognitiva funktionsnedsättningar som förknippas med sjukdomen hjälpa dem att utveckla strategier för att hantera dessa förändringar och för att upprätthålla en god relation med den drabbade personen. Bra information och råd finns att få från HS specialister och patientstödgrupper.Finns det strategier för att klara sjukdomen bättre?
Om du går till denna sida hittar du en lista över språksamordnare som kan hjälpa dig. Alternativt kan du använda kontaktformuläret som tillhandahålls här.Hur kontaktar jag EHDN?
Du kan boka en tid antingen genom att få en remiss från din husläkare eller genom att kontakta din lokala EHDN språksamordnare som kan hjälpa dig.Hur bokar jag tid hos en specialist?
Oberoende rådgivning om HS kan erhållas genom patientstödgrupperna i ditt land.Kan jag prata med en specialist utan att besöka ett sjukhus?
EHDN spelar en nyckelroll i den globala kliniska studien som heter Enroll-HD. Detta är en observationsstudie som inte inbegriper intervention vilket innebär att studien inte testar experimentella behandlingar. Deltagarna genomgår kliniska bedömningar vid årliga besök. Deltagare kan vara lämpliga att delta i kliniska prövningar av symtomatiska eller sjukdomsmodifierande behandlingar när dessa finns tillgängliga. ENROLL-HD finns tillgänglig vid många HS studieplatser runt om i världen. Vänligen titta här eller kontakta din EHDN språksamordnare, som kan informera dig om forskningsverksamhet i din region för att ta reda på om det finns en studieplats som är med i studien nära dig. Din lokala patientstödgrupp(er) kommer att kunna förse dig med allmän information om deltagande i forskning. Titta här för mer information om HS forskning eller besök webbsidan HDBuzz där forskare inom HS skriver om forskning på ett begripligt sätt (översatt till flera olika språk).Hur kan jag deltaga i forskning om HS?
Ja, det finns patientstödgrupper som ger stöd till individer och familjer som drabbats av HS. Dessa kan kontaktas via din allmänläkare eller en HS specialist eller om du väljer att själv kontakta dem direkt. European Huntington’s disease Association (EHA) har en lista över patientstödgrupper som kan vara användbar för dig.Finns det stödgrupper som är specialiserade på HS?
Vänligen kontakta din EHDN språksamordnare eller din lokala patientstödgrupp vid frågor.