HK Geschichte
Die HK ist benannt nach George Huntington, einem amerikanischen Arzt, der die Krankheit im Jahre 1872 beschrieb. Seine Darstellung basierte auf Beobachtungen von HK-betroffenen Familien aus dem Dorf East Hampton, Long Island, New York (USA), wo Dr. Huntington lebte und arbeitete. HK war in der Vergangenheit als Huntingtonsche Chorea und Veitstanz bekannt.
Symptome & Krankheitsverlauf
Die HK ist eine seltene Erkrankung, die 5 bis 10 pro 100.000 Menschen in der europäischen Bevölkerung betrifft. Eine ähnliche Prävalenz findet sich in Ländern, deren Populationen in erster Linie von europäischer Abstammung sind, wie den USA. Die HK ist in asiatischen und afrikanischen Ländern weniger verbreitet. Die Prävalenz wurde auf 1 pro 100.000 Menschen geschätzt. Männer und Frauen haben die gleiche Wahrscheinlichkeit, die HK-Expansion zu erben und die Krankheit zu entwickeln. Die HK zeichnet sich durch eine Kombination von motorischen (Bewegung), Verhaltens- (z.B. Stimmung) und kognitiven (z.B. Verständnis) Störungen aus, aber auch andere Symptome können berichtet werden. Die Symptome der HK können in Schwere, Alter beim Ausbruch und Grad des Fortschreitens von Individuum zu Individuum variieren – auch innerhalb der Mitglieder der gleichen Familie. Eine Person kann eine sehr offensichtliche Bewegungsstörung haben, aber nur leichte Verhaltenssymptome und kognitive Verschlechterung vorweisen, während eine andere jahrelang an Depressionen und Ängsten leiden kann, bevor sie irgendwelche abnormen Bewegungen vorweisen. Der Beginn der HK wird als „schleichend“ beschrieben, da es in der Regel schwierig ist, ein Datum zu bestimmen, wann sie tatsächlich beginnt. Die meisten Menschen, die HK-Mutationsträger sind, entwickeln Symptome im mittleren Erwachsenenleben – das heißt zwischen dem 35. und 55. Lebensjahr. Etwa 10% entwickeln Symptome vor dem 20. Lebensjahr (sie haben juvenile HK) und weitere 10% nach dem 55. Lebensjahr. Im Allgemeinen entwickelt sich die HK ganz allmählich, so dass die Diagnose eventuell erst nach vielen Jahren gestellt wird. Die durchschnittliche Krankheitsdauer beträgt 15 bis 20 Jahre ab Diagnosestellung, aber dies variiert zwischen den Individuen und kann auch von der Qualität der Versorgung abhängen, die der Patient erhält. Die Bestimmungsfaktoren für das Ausbruchsalter sind komplex und Gegenstand laufender Untersuchungen. Bei der Betrachtung großer Gruppen von HK-Patienten, fanden Wissenschaftler einen Zusammenhang zwischen der Trinukleotid-Wiederholungszahl und dem Alter bei Beginn der Symptome (Abbildung unten). Dies bedeutet im Allgemeinen, dass je höher die Anzahl der CAG-Wiederholungen ist, desto früher beginnen die Symptome (Abbildung unten). Jedoch kann für jede beliebige CAG-Wiederholungszahl die Variabilität des Alters bei Krankheitsbeginn bis zu 30 Jahre betragen, wie in der nachfolgenden Abbildung dargestellt wird. Dies ist vermutlich auf die Auswirkungen von anderen Genen als dem HTT (so genannte genetische Modifikatoren) und auf Umweltfaktoren, wie Lebensstil und Ernährung zurückzuführen. Zusammenfassend ist es sehr schwer das präzise Alter bei Krankheitsbeginn einer bestimmten Person, welche die HK-Expansion trägt, genau vorherzusagen. Entsprechend der Klassifizierung, entwickelt vom Neurologen und HK-Spezialisten Ira Shoulson von der Georgetown University in den USA, kann der Verlauf der HK in fünf Stufen unterteilt werden: Wenn die HK im frühen Lebensalter beginnt (vor dem 20. Lebensjahr), sind weniger unwillkürliche Bewegungen (Chorea) das markante Symptom, als verlangsamte Bewegungen (Bradykinesie) und Steifheit (Dystonie). Zu den frühen Merkmalen der juvenilen HK gehören deutliche Verhaltensänderungen, Schwierigkeiten beim Lernen und Sprechen, sowie ein Rückgang der schulischen Leistungen. Epileptische Anfälle werden gelegentlich berichtet, zumeist bei jungen Patienten. Im Allgemeinen verläuft die juvenile Form der Krankheit rapider als die erwachsene Form. Wenn die HK spät im Leben beginnt, ist Chorea meist ein markanteres Zeichen, als Langsamkeit oder Steifheit. In solchen Fällen ist es wahrscheinlich schwieriger, eine Familiengeschichte nachzuweisen, weil die Eltern der entsprechenden Person eventuell bereits gestorben sind, möglicherweise sogar bevor sie selbst Zeichen der Krankheit zeigten. Menschen mit der HK sterben nicht an den direkten Folgen der Krankheit, sondern eher auf Grund medizinischer Problemen, die sich aus dem geschwächten Zustand des Körpers ergeben. Dazu gehören Pneumonie (die für ein Drittel aller Todesfälle bei HK-Patienten verantwortlich ist), Ersticken, Herzversagen, Kopfverletzungen infolge von Stürzen und Ernährungsmangel. Das Selbstmordrisiko ist mit bis zu 7% aller Todesfälle der Patienten deutlich erhöht.Wie häufig ist die HK?
Was sind die Symptome der HK?
Die häufigsten psychiatrischen Symptome der HK sind Apathie, Angst, Depression, Reizbarkeit, Wutausbrüche, Impulsivität, Zwangsstörungen, Schlafstörungen und sozialer Rückzug. Seltener wird von Manie und Schizophrenie – einschließlich Wahnvorstellungen (realitätsfremde Vorstellungen) und Halluzinationen (sehen, hören oder fühlen von Dingen, welche nicht wirklich existieren) – berichtet. Betroffene Personen können Selbstmordgedanken haben, besonders in frühen Stadien der Krankheit. Die meisten HK-Patienten und ihre Betreuer nehmen die Verhaltens-Symptome, als beunruhigender wahr als die motorischen oder kognitiven Beeinträchtigungen, die durch die Krankheit verursacht werden.
Die HK zeichnet sich durch die allmähliche Beeinträchtigung von Verständnis, logische Denken, Urteilsvermögen und Gedächtnis aus. Kognitive Symptome beinhalten verlangsamtes Denken und Schwierigkeiten sich zu konzentrieren, zu organisieren, zu planen, Entscheidungen zu treffen und Fragen zu beantworten, sowie Kurzzeitgedächtnis- und Problemlösungsdefizite und eine beeinträchtigte Fähigkeit neue Informationen aufzunehmen und zu verstehen.
Es gibt eine Reihe weiterer Veränderungen, die während des Verlaufs der HK auftreten können, einschließlich Appetitlosigkeit, Gewichtsverlust, Verlust des Selbstwertgefühls, Verlust des Sexualtriebs, sowie Harn- und Stuhlinkontinenz.Wann treten die Symptome der HK auf?
Wodurch wird das Alter bei Symptombeginn bestimmt?
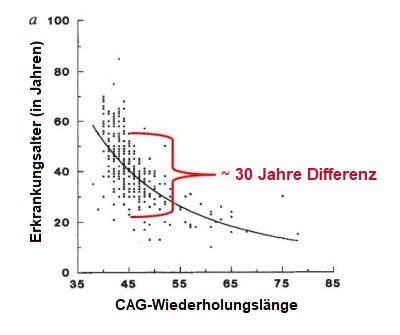
Nature Genetics 4, 398-403 (1993).Was sind die einzelnen Stadien des Verlaufs der HK?
Unterscheiden sich die Symptome der juvenilen HK von denen der
erwachsenen Form der HK?
Welche Symptome zeigen sich, wenn die HK spät im Leben auftritt?
Todesursachen
Diagnose & Behandlung
Die HK wird durch eine Kombination aus klinischen Untersuchungen und einem genetischen Test diagnostiziert. Die klinische Diagnostik basiert auf der medizinischen und familiären Geschichte einer Person, sowie auf Standarduntersuchungen, die mit Hilfe von klinischen Bewertungsskalen die Beurteilung der Häufigkeit und Schwere der Symptome der HK beurteilen. Die Ergebnisse der klinischen Diagnostik werden in der Regel durch genetisches Screening für die HTT-Expansion (bekannt als diagnostische oder bestätigende Gentests) bestätigt. Wenn eine Person keine Symptome zeigt, sondern ein Risiko für die Erkrankung hat, wird ein asymptomatischer Gentest (bekannt als prädiktiver Gentest) gemacht um zu bestimmen, ob sie die Expansion trägt oder nicht.Wie wird die HK diagnostiziert?
Die klinischen Beurteilungsmethoden, die zur Diagnose der HK und zum Messen der Aspekte der Darstellung genutzt werden, sind nicht in allen Kliniken in allen Ländern einheitlich. Allerdings ist die am häufigsten verwendete Methode, die Unified Huntington’s Disease Rating Scale (UHDRS), die in motorische, verhaltensbezogene, kognitive und funktionale Teilbereiche aufgeteilt ist. Darüber hinaus wird das Problem Behaviors Assessment for Huntington’s Disease (PBA) häufig für die Beurteilung der Schwere und Häufigkeit von Verhaltensstörungen (wie depressive Stimmung, Apathie und Reizbarkeit) benutzt, während eine Vielzahl von Tests wie die Mini-Mental State Examination (MMSE) und die Mattis Dementia Rating Scale verwendet werden, um den Teilbereich des UHDRS zu ergänzen, welcher kognitive Einschränkungen bewertet.Welche klinischen Bewertungsinstrumente werden zur Diagnose der HK verwendet?
Mit dem Wissen zu leben, dass man Risikoperson für die HK ist, kann sehr besorgniserregend sein. Es kann sein, dass Sie es vorziehen würden, sicher zu wissen, ob Sie die Expansion tragen. In diesem Fall sind genetische Beratung und psychologische Unterstützung dringend zu empfehlen, da diese es Ihnen erlauben, Ihre Möglichkeiten zu erkunden und Ihre Sorgen zu besprechen. Im Allgemeinen werden prädiktive Tests nicht für diejenigen empfohlen, die jünger als 18 Jahre alt sind – das Alter, in dem man hofft, dass eine Person die Reife hat, mit dem Wissen umzugehen, dass sie die Expansion trägt. In Ausnahmefällen kann es sich jedoch als vernünftig erweisen, den bestätigenden Gentest bei Kindern durchzuführen – wenn sie beispielsweise Anzeichen von jugendlicher HK zeigen – oder bei Frauen, die jünger sind als 18, wenn sie schwanger sind. Wenn Sie sich dafür entscheiden getestet zu werden, wird eine Blutprobe aus einer Vene in Ihrem Arm entnommen und dauraus wird im Labor die DNA extrahiert. Je nach lokalem Service ist das Ergebnis in zwei bis acht Wochen vorhanden. Die Richtlinien für das prädiktive genetische Testverfahren wurde von der EHDN Genetic Testing und Counselling Working Group im Jahr 2012 aktualisiert.Wie ist das Verfahren für prädiktive Gentests?
Der genetische Test ermittelt die Anzahl der CAG – Wiederholungen im HTT – Gen. Der Test kann zeigen, ob Sie die HK – Expansion tragen, aber er kann nicht feststellen, wann die Krankheit ausbrechen wird, wie schnell sie voranschreiten wird oder welche Symptome Sie entwickeln könnten. Der genetische Test für HK gilt als nahezu 100% genau. Die Ergebnisse aus der DNA-Analyse werden in der Regel mit zwei separaten Blutproben doppelt kontrolliert. Darüber hinaus könnte auch Blut von einem Elternteil der betroffenen Person (oder, falls dies nicht verfügbar ist, von einem anderen Familienmitglied) geprüft werden, um eine Bestätigung der ursprünglichen Diagnose zu liefern.Was stellt der genetische Test fest?
Ja. Dies kann durch ein modernes diagnostisches Verfahren, der Genetische Präimplantationsdiagnostik (PGD) – auch Embryoscreening genannt – erreicht werden, das in Kombination mit In-vitro-Fertilisation (IVF) verwendet wird und das Screening von Embryonen vor der Implantation im Mutterleib beinhaltet. Die Technik stellt sicher, dass nur Embryonen, welche normale Kopien des Gens geerbt haben, implantiert werden. Daher macht es die PGD möglich, auch wenn einer von ihnen die HK-Mutation trägt, dass ein Paar ein Kind empfangen kann, das nicht das mutierte HTT-Gen trägt – unabhängig davon, ob der Mann oder die Frau Träger ist. Allerdings ist die PGD nach dem Embryonenschutzgesetzen in einigen Ländern nicht zulässig und es ist auch wichtig zu beachten, dass die Chancen einer Schwangerschaft nach einer PGD / IVF niedriger sind als im Fall einer „natürlichen“ Empfängnis. In einigen Ländern ist es möglich ungeborene Föten zu testen, welche auf natürlichem Wege empfangen wurden, und sich eventuell für einen Schwangerschaftsabbruch zu entscheiden, sobald der genetische Status des Fötus bekannt ist.Ist es für eine Person, welche die HK-Expansion trägt, möglich
sicherzustellen, dass sie die HK nicht an ihr Kind weitergibt?
Eine pränatale Diagnose (vor der Geburt) kann nur durchgeführt werden, wenn diejenigen, die eine solche Untersuchung wünschen auch belegen können, dass ihr Fall bestimmte medizinische und rechtliche Kriterien erfüllt. Diese können von Land zu Land unterschiedlich sein. Es gibt zwei Standardverfahren für die pränatale Diagnostik. Das erste ist die Amniozentese (auch als Fruchtwasseruntersuchung bezeichnet). Hier wird Fruchtwasser, welches fetale Zellen enthält, über eine Nadel, die durch die Bauchwand der Mutter eingeführt wird, entnommen – für gewöhnlich wird diese Untersuchung nach der 14. Schwangerschaftswoche vorgenommen. Das zweite ist die Chorionzottenbiopsie, bei der eine Probe von Chorionzotten (Plazentagewebe) entnommen wird und die früher durchgeführt werden kann – zwischen der 10. und 13. Schwangerschaftswoche. Diese ist jedoch risikoreicher für den Fötus.Kann ich mein ungeborenes Kind testen lassen?
Es gibt derzeit keine Therapien, die die zugrunde liegenden Ursachen der HK effektiv behandeln können. Allerdings wurde durch Grundlagenforschung und klinische Forschung in den letzten Jahren das Wissen über die HK bedeutend erhöht und viele , welche die Krankheitsentstehung untersuchen, um Medikamente zu finden, die den Krankheitsbeginn verschieben oder das Fortschreiten verlangsamen könnten. Behandlungen, die bestimmte Symptome der Krankheit (symptomatische Behandlungen) lindern und somit die Lebensqualität der Patienten verbessern gibt es bereits. Diese werden in pharmakologische (medikamentöse) und nicht-pharmakologische (nicht-medikamentöse) Behandlungen unterteilt.Gibt es Behandlungen bei HK?
Chorea, Bradykinesie, Reizbarkeit, Apathie, Depression, Angst und Schlafstörungen wurden als am meisten belastende Symptome der HK beschrieben. Es gibt mehrere Möglichkeiten für diese Symptome mit Medikamenten zu regulieren. Allerdings können viele der Medikamente Nebenwirkungen verursachen und einige können den therapeutischen Wirkungen anderer Medikamente entgegenwirken. Darüber hinaus kann das gleiche Medikament unterschiedliche Effekte bei verschiedenen Personen haben. Die Behandlung muss durch erfahrene HK-Spezialisten entsprechend der Symptome des Patienten und seiner Reaktion auf die betreffenden Medikamente angepasst werden.Welche medikamentösen Möglichkeiten für die Behandlung von HK-Symptomen gibt es?
Nicht-medikamentöse Behandlungen (wie Psychotherapie und kognitive, physische, Sprach-, Atem- und Ergotherapie) können die psychologischen und physischen Symptome der HK verbessern. Zum Beispiel wurden von Verbesserungen der Stimmung, der motorischer Steuerung, der Sprache, des Gleichgewichts, des Schluckens und des Gangs nach diesen Therapien berichtet. Es ist bekannt, dass körperliche Bewegung sowohl die körperliche als auch die psychische Gesundheit und das allgemeine Wohlbefinden verbessern können. Es wurde gezeigt, dass durch Training die Symptome einer Depression gelindert werden können. Es gibt eine Vielzahl von Beweisen dafür, dass es auch dazu beitragen kann, das Fortschreiten der Bewegungsbeeinträchtigung bei der HK zu verlangsamen. Zum Beispiel haben einige Physiotherapie-Programme gezeigt, dass sie sich begünstigend auf motorische Symptome, de Gang und das Gleichgewicht. Die EHDN Physiotherapie-Arbeitsgruppe hat einen Leitfaden für Physiotherapeuten veröffentlicht, welche mit HK-Patienten arbeiten.Wie können nicht-medikamentöse Behandlungen helfen?
Die Vorteile einer Diät reich an Vitaminen, Coenzymen und anderen Wirkstoffen, waren Gegenstand vieler Diskussionen, sind aber klinisch noch nicht nachgewiesen. Da Gewichtsverlust ein Problem für einige HK-Patienten, vor allem in den späteren Stadien der Krankheit ist, ist es wichtig, eine gesunde Ernährung im Laufe der Krankheit zu gewährleisten. In späteren Stadien kann eine kalorienreiche Diät notwendig werden. Die Überweisung an einen Ernährungsberater kann daher hilfreich sein.Kann eine spezielle Diät die Symptome der HK lindern?
Vererbung & Ursache der HK
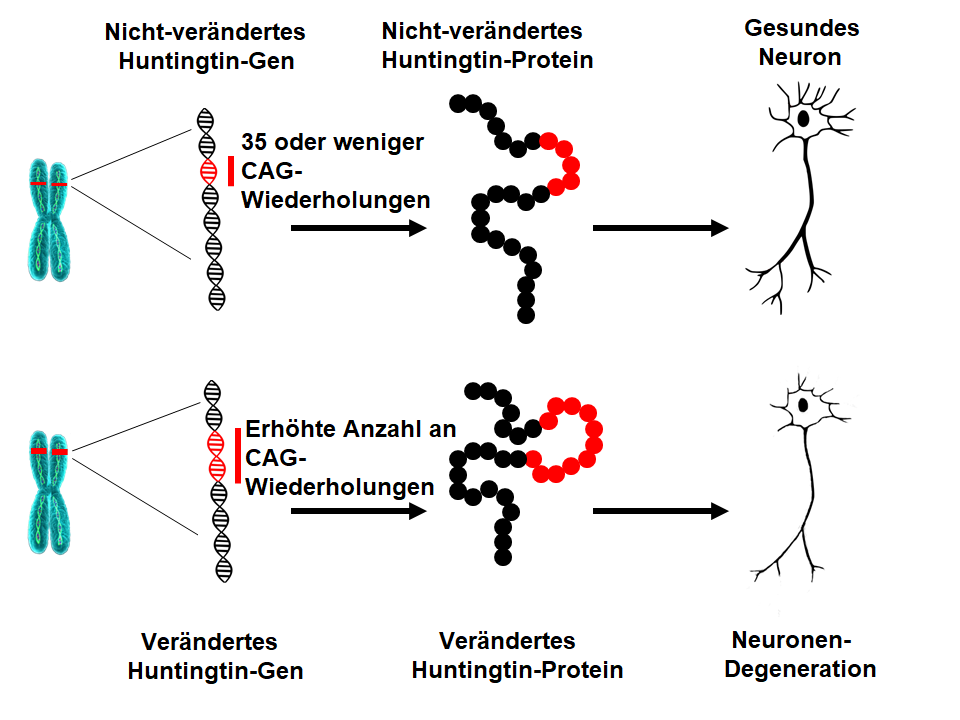
Die HK wird durch eine Veränderung (eine Expansion) im Gen (HTT) verursacht, welches ein Protein namens Huntingtin codiert. Als Ergebnis dieser Expansion wird das Gen in eine veränderte Form des Proteins übersetzt, was eine Fehlfunktion und das Absterben von Nervenzellen (Neuronen) in bestimmten Bereichen des Gehirns zur Folge hat. Die genauen Mechanismen der Erkrankung sind vielfältig und hochkomplex, in Übereinstimmung mit den multiplen Funktionen des Huntingtin-Proteins. Die Forscher arbeiten daran, die zugrundeliegenden krankheitsverursachenden Mechanismen für die Entwicklung von krankheitsmodifizierenden Therapien besser zu verstehen.Wodurch wird die HK verursacht?
Im Jahr 1993 identifizierten Wissenschaftler die Mutation, welche die HK verursacht. Das HTT-Gen befindet sich auf dem Chromosom 4 und kodiert ein Protein namens Huntingtin. Das Gen enthält eine Sequenz von drei Nukleotiden (die Grundeinheiten der DNA), Cytosin-Adenin-Guanin (CAG), die mehrmals wiederholt wird. Diese sogenannte Trinucleotidwiederholung kann in der Länge variieren. Wenn eine Person 40 CAG-Wiederholungen oder mehr in einer Kopie des HTT-Gens hat, wird diese die HK innerhalb einer normalen Lebensdauer entwickeln – das heißt im mittleren Erwachsenenalter. Da die Expansion, welche die HK verursacht in allen Zellen des Körpers seit der Zeugung vorliegt und an nachfolgende Generationen weitergegeben werden kann, ist die HK eine erbliche Erkrankung.Was ist die zugrundeliegende Ursache für die HK?
Je höhrer die Anzahl der CAG-Wiederholungen ist, desto instabiler wird dieser bestimmte Abschnitt der DNA. Dies bedeutet, dass sich die Anzahl der Wiederholungen in diesem Abschnitt erhöhen oder verringern kann, wenn sie an die nächste Generation weitergegeben wird. Solange die Anzahl der CAG-Wiederholungen im HTT-Gen niedriger als 27 ist, ist der Abschnitt stabil. Wenn die Anzahl der Wiederholungen zwischen 27 und 35 liegt (die sogenannte intermediäre Wiederholungslänge), wird man die HK nicht entwickeln und der Abschnitt gilt als normal. Allerdings ist eine CAG-Wiederholungszahl von 27 oder mehr instabil und kann sich erhöhen, wenn sie an die nächste Generation weitergegeben wird. Das bedeutet, dass diese Kinder das Risiko haben, die HK zu entwickeln. Personen mit einer CAG-Wiederholungszahl zwischen 36 und 39 können die HK entwickeln aber wenn überhaupt, dann nur sehr spät im Leben. Hierbei spricht man vom Bereich der verminderten Penetranz. Wenn die Anzahl der CAG-Wiederholungen höher als 39 ist, wird man die HK innerhalb einer normalen Lebensdauer entwickeln – meistens im mittleren Erwachsenenleben. In seltenen Fällen kann die CAG-Expansion außergewöhnlich lang sein, was zu einem Krankheitsbeginn im Jugendalter oder Kindesalter führt (juvenile HK). Patienten, die die Krankheit vor dem 10. Lebensjahr entwickeln, haben oft mehr als 80 CAG-Wiederholungen.Welche Bedeutung hat die Länge der HTT CAG-Wiederholung?
CAG-Wiederholungslänge
Ursache für die Krankheit?
Auswirkungen auf die Nachkommen?
Name
Unter 27
Nein
Keine
Normale Wiederholungslänge
27-35
Nein
"Wiederholungen von 27 und mehr können instabil sein und könnten sich erhöhen wenn sie an die Nachkommen weitergegeben werden"
Intermediäre Wiederholungslänge
36-39
Vielleicht
Ja, Nachkommen werden mit einer 50%igen Wahrscheinlichkeit das verlängerte Gen erben.
Bereich der verminderten Penetranz
40 und darüber
Ja
Ja, Nachkommen werden mit einer 50%igen Wahrscheinlichkeit das verlängerte Gen erben.
Wiederholungslänge mit voller Penetranz
Gene finden sich auf unseren Chromosomen in jeder Zelle unseres Körpers. Ein Gen ist ein DNA-Abschnitt, welcher den Code für ein bestimmtes Protein enthält; Die DNA wird in die Messenger-RNA (mRNA) umgeschrieben, die dann in das Protein übersetzt wird. In der Regel erbt jeder zwei Kopien eines jeden Gens – eine von der Mutter, eine vom Vater. Bei der HK ist das wichtige Gen das HTT-Gen, welches das Huntingtin-Protein kodiert. Wenn ein Kind eine erweiterte Version des HTT-Gens erbt, dann wird dieses Kind die HK ebenfalls entwickeln. Der Elternteil selbst kann bereits die Krankheit haben oder sie kann sich später entwickeln.Was ist ein Gen?
Proteine sind große Moleküle aus Bausteinen namens Aminosäuren. Die genaue Sequenz von Aminosäuren in einem bestimmten Protein wird durch die DNA-Sequenz des entsprechenden Gens bestimmt. Gene fungieren daher als Vorlagen – als Sätze von Anweisungen für Zellen, die ihnen sagen, wie man spezifische Proteine baut. Das HTT-Gen enthält Anweisungen zum Aufbau des Huntingtin-Proteins. Proteine sind die Moleküle, die die Arbeit innerhalb der Zellen durchführen – sie führen eine große Anzahl von wesentlichen Prozessen wie Enzymreaktionen oder Unterstützung der Zellstruktur durch. Wenn ein Protein aufgrund einer Expansion des Gens das es codiert abnormal funktioniert oder fehlt, dann kann dies die Zelle und letztlich den ganzen Organismus beeinflussen, was manchmal eine Krankheit verursacht.Was ist ein Protein?
Das Huntingtin-Protein ist ein sehr großes Protein, das in jeder Zelle des menschlichen Körpers in unterschiedlichem Maße hergestellt oder „exprimiert“ wird; die höchsten Konzentrationen werden im Gehirn gefunden. Huntingtin scheint ein sehr wichtiges Protein zu sein, da sein Fehlen für Mausembryonen tödlich ist. An einem bestimmten Ende des Huntingtin-Proteins gibt es einen Abschnitt von Wiederholungen einer bestimmten Aminosäure namens Glutamin. Diese unverwechselbare Eigenschaft, bekannt als die Polyglutamin-Wiederholung, besteht normalerweise aus bis zu 35 Glutamin-Einheiten. Bei Personen, die die HK-Expansion tragen, enthält sie jedoch mindestens 36 Wiederholungen und es ist diese Polyglutamin-Expansion, die zu einem falsch funktionierendem Protein führt.Das Huntingtin Protein
Die HK ist eine dominante Erbkrankheit. Dies bedeutet, dass eine Person die mit einer Kopie des mutierten HTT-Gens geboren wird, die HK entwickeln wird, obwohl sie auch eine normale Kopie des Gens trägt. Ein Träger der HK-Expansion, ob symptomatisch oder nicht, kann entweder eine normale oder eine mutierte Kopie jedes Gens mit einer 50% igen Wahrscheinlichkeit weitergeben (vorausgesetzt, dass er die Expansion nur auf einer der beiden Kopien des HTT-Gens trägt ). Medizinische Techniken können sicherstellen, dass eine betroffene Person nur das normale HTT-Gen an seine Kinder weitergibt. Auf der anderen Seite wird eine Person, die das mutierte HTT-Gen nicht geerbt hat, die Krankheit nicht entwickeln, und auch seine/ihre Kinder sind nicht gefährdet. Die HK-Expansion kann keine Generation überspringen. Allerdings kann es passieren, dass ein Expansionsträger stirbt, bevor er Symptome zeigt, und dass seine oder ihre Kinder nicht wissen, dass sie das Risiko tragen, die Krankheit zu entwickeln.Wie wird die HK vererbt?
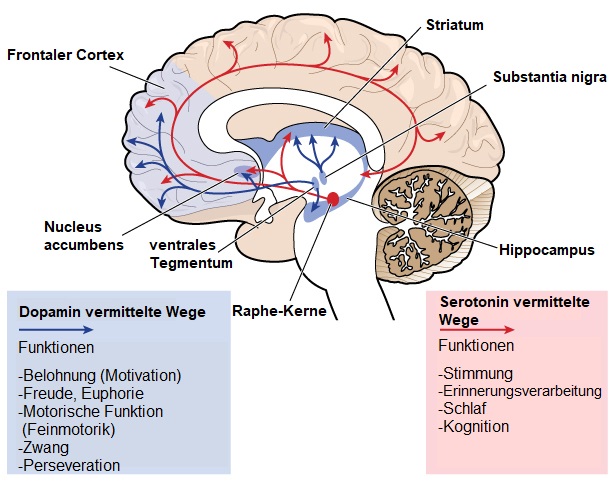
Bestimmte Funktionen des Gehirns wie die Fähigkeit sich zu bewegen, zu denken und zu reden werden bei der HK allmählich schlechter, da entscheidende Nervenzellen beschädigt werden und sterben. Der Teil des Gehirns, der am meisten von der HK betroffen ist, ist das Striatum, das Bestandteil der Basalganglien ist und sich tief in der zentralen Region des Gehirns befindet. Das Striatum ist in erster Linie an der Planung und Steuerung von Bewegungen beteiligt aber auch an vielen anderen Prozessen, einschließlich Kognition und Emotionen. Während die HK fortschreitet, beeinflusst sie den Kortex (der äußerste faltige Teil des Gehirns), was zur kognitiven Verschlechterung beiträgt. Im Allgemeinen verursacht die HK im Laufe der Zeit eine Atrophie des gesamten Gehirns und beeinträchtigt die allgemeine Funktionsfähigkeit.Von der HK betroffene Gehirnareale
HK im täglichen Leben
Positiv auf für die HK-Expansion getestet zu sein kann viele verschiedene Aspekte des Lebens eines Menschen beeinflussen, einschließlich der Entscheidung, ob man Kinder haben möchte, Pläne für die Zukunft zu machen, Prioritäten zu überdenken, eine passende Wohnung zu suchen und andere Familienmitglieder darüber in Kenntnis zu setzen, dass sie auch von der Krankheit betroffen sein könnten. Vor allem junge Erwachsene müssen die Auswirkungen eines positiven Testergebnisses auf ihre Aus-, Fortbildung und ihr Berufsleben bedenken. Wenn die Krankheit fortschreitet, beeinflusst sie allmählich die Fähigkeit einer Person, unabhängig zu leben. Die Arbeit, das soziale Leben und allgemeine tägliche Aktivitäten werden problematisch und Patienten werden zunehmend von der Hilfe von Angehörigen und Gesundheits- und Sozialpflegern abhängig. Lokale Patientenselbsthilfegruppen und klinische Zentren können jederzeit kontaktiert werden und bieten Unterstützung an.Wie beeinflusst die HK das tägliche Leben?
Effiziente Strategien für die Bewältigung der HK müssen individuell abgestimmt sein und hängen von der betroffenen Person, dem Stadium der Krankheit und dem familiären Umfeld ab. Die HK entwickelt sich sehr langsam, so dass für gewöhnlich Zeit bleibt, sich an die Veränderungen anzupassen, welche die Krankheit mit sich bringt. Für Betreuer und Angehörige kann ein besseres Verständnis der Verhaltens- und kognitiven Beeinträchtigungen, die mit der Krankheit verbunden sind helfen Strategien zu entwickeln, um sich diesen Veränderungen anzupassen und eine gute Beziehung mit der betroffenen Person aufrechtzuerhalten. Nützliche Informationen und Ratschläge erhalten Sie von HK-Spezialisten und Patientenselbsthilfegruppen.Gibt es irgendwelche Strategien um besser mit der HK umzugehen?
Wenn Sie auf diese Seite gehen, finden Sie eine Liste der Sprachbereichskoordinatoren, die Ihnen helfen können. Alternativ können Sie auch das hier hinterlegte Kontaktformular verwenden.Wie kann ich das EHDN kontaktieren?
Sie können einen Termin vereinbaren, indem Sie entweder eine Überweisung von Ihrem Hausarzt erhalten, oder indem Sie Ihren lokalen EHDN lokalen EHDN Sprachbereichskoordinator kontaktieren, der Ihnen helfen wird.Wie bekomme ich einen Termin mit einem Fachmann?
Unabhängige Beratung über die HK können Sie von den Patientenselbsthilfegruppen in Ihrem Land erhalten.Gibt es irgendeine Art, wie ich mit einem Spezialisten sprechen kann
ohne eine Klinik zu besuchen?
Das EHDN spielt eine Schlüsselrolle in der weltweiten klinischen Studie Enroll-HD. Dies ist eine Beobachtungsstudie, die keine Eingriffe beinhaltet. Das bedeutet, dass per se keine experimentellen Therapien an Ihnen getestet werden. Die Teilnehmer werden während ihrer jährlichen Besuche klinisch beurteilt und können an klinischen Studien mit symptomatischen oder krankheitsverändernden Behandlungen teilnehmen, sobald diese verfügbar sind. Enroll-HD ist über viele HK- Studienzentren rund um die Welt erreichbar. Um herauszufinden, ob es einen Standort in Ihrer Nähe gibt, schauen Sie bitte hier oder kontaktieren Sie ihren EHDN- Sprachbereichskoordinator der Sie über die Forschungsaktivitäten in Ihrer Region informieren kann. Ihre regionalen Patientenselbsthilfegruppe(n) können Ihnen allgemeine Informationen über die Teilnahme an der Forschung geben. Für weitere Informationen über die HK-Forschung, schauen Sie bitte hier oder besuchen Sie die HDBuzz-Webseite mit HK-Forschungsnachrichtigungen, die von HK-Forschern in Laiensprache geschrieben und in die meisten Sprachen übersetzt.Wie kann ich mich an der HK Forschung beteiligen?
Ja, eine Reihe von Patientenselbsthilfegruppen bieten Unterstützung für Einzelpersonen und Familien an, die von der HK betroffen sind. Diese können über Ihren Hausarzt oder HK-Spezialisten kontaktiert werden oder Sie können direkt mit ihnen Kontakt aufnehmen. Die European Huntington’s disease Association (EHA) hält eine Liste der Patientenselbsthilfegruppen bereit, die für Sie nützlich sein könnten.Gibt es Selbsthilfegruppen, die auf die HK spezialisiert sind?
Für weitere Fragen wenden Sie sich bitte an Ihren EHDN- Sprachbereichskoordinator oder Ihre lokale Patientenselbsthilfegruppe.