Histoire de la MH
La MH tient son nom de George Huntington, un médecin américain qui a décrit la maladie en 1872. Sa description était fondée sur l’observation de familles affectées par la MH issues d’un village de l’East Hampton, de Long Island et de New York (USA), où le Dr Huntington a vécu et travaillé. La MH était connue par le passé en tant que chorée de Huntington et Danse de Saint Guy.
Symptômes et progression de la maladie
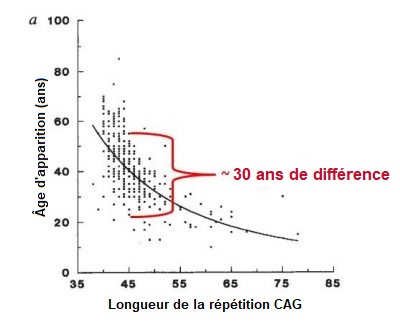
La MH est une maladie rare qui affecte 5 à 10 personnes sur 100,000 dans la population Européenne. On retrouve une prévalence similaire dans les pays dont la population descend principalement des Européens, comme par exemple les Etats-Unis. La MH est moins répandue dans les pays Asiatiques et Africains, où la prévalence a été estimée à 1 personne sur 100,000. Les hommes et les femmes courent le même risque d’hériter de l’expansion MH et de développer la maladie. La MH se caractérise par une combinaison de perturbations motrices (mouvement), comportementales (par exemple l’humeur) et cognitives (par exemple la compréhension), mais d’autres symptômes peuvent également être rapportés. La sévérité des symptômes de la MH, l’âge d’apparition et la vitesse de progression peuvent varier d’un individu à l’autre – ainsi qu’entre les membres d’une même famille. Une personne peut avoir des troubles du mouvement très remarquables mais des symptômes comportementaux et une déterioration cognitives modérés, tandis qu’une autre peut souffrir de dépression et d’anxiété pendant des années avant de manifester des mouvements anormaux. L’apparition de la MH est décrite comme « insidieuse », car c’est généralement difficile de déterminer une date réelle d’apparition. La plupart des individus porteurs de la mutation MH développent des symptômes en milieu de vie adulte – c’est-à-dire entre 35 et 55 ans. Dans 10% des cas, cela se produit avant l’âge de 20 ans (il s’agit de la forme juvénile de la MH), et pour d’autres 10% après l’âge de 55 ans. En général, la MH se développe très graduellement, et peut rester non diagnostiquée pendant plusieurs années. En moyenne, la durée de la maladie est de 15 à 20 ans à partir du diagnostic, mais cela varie entre individus et peut aussi dépendre de la qualité de la prise en charge que le patient reçoit. Les déterminants de l’âge d’apparition sont complexes et font l’objet de recherches en cours. En regardant de larges groupes de patients MH, les scientifiques trouvent une corrélation entre le nombre de répétitions du trinucléotide et l’âge d’apparition des symptômes (figure ci-dessous). Cela signifie que, en général, plus le nombre de répétitions CAG est élevé, plus tôt les symptômes apparaîtront (figure ci-dessous). Toutefois, pour un nombre de répétitions CAG donné, la variabilité de l’âge d’apparition peut aller jusqu’à 30 années, comme montré sur la figure ci-dessous. C’est probablement dû aux effets de gènes autres que celui de la HTT (appelés modificateurs génétiques), et à des facteurs environnementaux tels que le mode de vie et le régime alimentaire. En tenant compte de tout cela, il est très difficile de prédire avec précision l’âge d’apparition chez une personne donnée porteuse de la mutation MH. Selon une classification développée par le neurologue et spécialiste de la MH Ira Shoulson de l’Université de Georgetown aux Etats-Unis, la progression de la MH peut se diviser en cinq stades : Lorsque la MH débute tôt dans la vie (avant l’âge de 20 ans), les mouvements involontaires (chorée) sont moins marqués que la lenteur des mouvements (bradykinésie) et la raideur (dystonie). Les caractéristiques précoces de la MH juvénile incluent des changements comportementaux prononcés, des difficultés d’apprentissage et d’élocution, et un déclin des performances scolaires. En général, la forme juvénile de la maladie progresse plus rapidement que la forme adulte. Lorsque la MH débute tardivement dans la vie, la chorée a tendance à être plus marquée que la lenteur ou la rigidité. Dans de tels cas, il est souvent plus difficile d’établir les antécédents familiaux de la personne, car ses parents peuvent être déjà décédés, peut-être même avant d’avoir développé les symptômes de la maladie. Les personnes atteintes de MH ne meurent pas directement de la maladie, mais plutôt de complications médicales qui résultent de l’affaiblissement de l’organisme. Elles incluent les pneumonies (qui représentent un tiers des décès chez les patients MH), les fausses routes, les blessures à la tête consécutives à des chutes, ainsi que les déficits nutritionnels. Le risque de suicide est notablement augmenté, représentant jusqu’à 7% des décès chez ces patients.La MH est-elle une maladie courante ?
Quels sont les symptômes de la MH ?
Les symptômes psychiatriques les plus fréquents dans la MH sont l’apathie, l’anxiété, la dépression, l’irritabilité, les accès de colère, l’impulsivité, les comportements obsessionnels-compulsifs, les perturbations du sommeil et l’isolement social. Plus rarement, on rapporte des épisodes de manie et schizophrénie, incluant des délires (fausses croyances) et des hallucinations (visuelles, auditives ou sensations de choses qui n’existent pas réellement). Les individus affectés peuvent connaître des pensées suicidaires, particulièrement au cours des stades précoces de la maladie. La plupart des patients MH et leurs aidants perçoivent les symptômes comportementaux comme étant plus pénibles que les détériorations motrices ou cognitives causées par la maladie.
La MH se caractérise par la détérioration graduelle de la compréhension, du raisonnement, du jugement et de la mémoire. Les symptômes cognitifs incluent un ralentissement de la pensée et des difficultés de concentration, d’organisation, de planification, de prise de décision et de la réponse aux questions, ainsi qu’un déficit de la mémoire à court terme et de la résolution de problèmes et une réduction de la capacité à saisir et comprendre de nouvelles informations.
Il existe un certain nombre d’autres changements pouvant survenir au cours de la progression de la MH, comme la perte d’appétit, la perte de poids, la perte de confiance en soi, la perte de libido et l’incontinence urinaire et fécale.Quand les symptômes de la MH apparaissent-ils ?
Qu'est-ce qui détermine l'âge d'apparition des symptômes ?
Nature Genetics 4, 398–403 (1993).Quels sont les différents stades de progression de la MH ?
peut fonctionner normalement, tant au domicile qu’au travail.
une capacité amoindrie. Il/elle est encore capable d’accomplir les tâches
de la vie quotidienne mais éprouve quelques difficultés.
les responsabilités domestiques. Il/elle a besoin d’aide ou de surveillance
importante pour pouvoir gérer ses comptes. Les autres activités
quotidiennes peuvent être quelque peu difficiles, mais le plus souvent ne
nécessitent qu’une aide mineure.
mais est encore capable de vivre chez elle avec l’aide de sa famille ou
de personnel d’accompagnement.
activités de la vie quotidienne et des soins infirmiers sont nécessaires.Les symptômes de la MH juvénile sont-ils différents de ceux de la
forme adulte ?Quels sont les symptômes de la MH lorsque celle-ci débute
tardivement dans la vie ?Causes du décès
Diagnostic & Traitement
La MH est diagnostiquée par une combinaison d’évaluations cliniques et un test génétique. Le diagnostic clinique est basé sur l’histoire médicale et familiale de la personne, mais aussi sur des examens standards qui utilisent des échelles de cotation cliniques qui évaluent la fréquence et la sévérité des symptômes de la MH. Les résultats du diagnostic clinique sont généralement confirmés par une analyse génétique de l’expansion HTT (appelé diagnostic ou test génétique de confirmation). Si une personne ne manifeste aucun symptôme, mais est à risque pour la maladie, un test génétique asymptomatique (appelé test génétique prédictif) déterminera si elle est porteuse ou non de l’expansion.Comment la MH est-elle diagnostiquée ?
Les outils d’évaluation clinique utilisés pour diagnostiquer la MH et mesurer les aspects de la présentation ne sont pas les mêmes dans tous les hôpitaux de tous les pays. Toutefois, l’outil le plus fréquemment utilisé est le Unified Huntington’s Disease Rating Scale (UHDRS), qui comprend les sous-sections motrice, comportementale, cognitive et fonctionnelle. De plus, le Problem Behaviours Assessment for Huntington’s Disease (PBA) est souvent utilisé pour évaluer la sévérité et la fréquence des anomalies comportementales (telles que l’humeur dépressive, l’apathie et l’irritabilité), tandis qu’une variété de tests tels que le Mini-Mental State Examination (MMSE) et le Mattis Dementia Rating Scale sont utilisés pour compléter la sous-section de l’UHDRS qui évalue les détériorations cognitives.Quels sont les outils d'évaluation clinique utilisés pour diagnostiquer
la MH ?
Vivre en sachant que vous êtes à risque pour la MH peut être très préoccupant. Vous pouvez avoir le sentiment de préférer être certain d’être porteur de la mutation. Dans ce cas, un conseil en génétique et un soutien psychologique sont hautement recommandés, car ils vous permettent d’explorer les options et de discuter de vos préoccupations. En général, le test prédictif n’est pas recommandé pour les moins de 18 ans – car on peut penser qu’à partir de cet âge une personne a assez de maturité pour faire face à la connaissance de son statut de porteur. Toutefois, dans certains cas exceptionnels, il peut être raisonnable d’effectuer le test génétique de confirmation chez des enfants – s’ils montrent des signes de MH juvénile, par exemple – ou chez des jeunes femmes de moins de 18 ans si elles sont enceintes. Si vous décidez de faire le test, un échantillon de sang sera prélevé à partir d’une veine de votre bras et votre ADN en sera extrait au laboratoire. En fonction des procédures locales, le résultat sera disponible d’ici 2 à 8 semaines. Les recommandations pour la procédure de test génétique prédictif ont été mises à jour par le Groupe de Travail sur le Test et Conseil Génétique d’EHDN en 2012.Quelle est la procédure du test génétique prédictif ?
Le test génétique détermine le nombre de répétitions de CAG sur le gène HTT. Le test peut dire si vous êtes porteur de la mutation MH, mais ne peut pas dire quand la maladie débutera, quelle sera sa vitesse de progression, ou quels symptômes vous pourriez développer. On considère que la fiabilité du test génétique pour la MH est proche de 100%. Les résultats de l’analyse de l’ADN sont généralement vérifiés deux fois en utilisant deux échantillons de sang distincts. De plus, on peut également tester le sang d’un parent de la personne affectée (ou, si c’est impossible, d’un autre membre de sa famille) pour confirmer le diagnostic d’origine.Que détecte le test génétique ?
Oui. C’est faisable grâce à une procédure de diagnostic moderne appelée diagnostic génétique pré-implantatoire (DPI) – aussi appelée sélection embryonnaire – utilisée en association avec la fécondation in vitro (FIV), et qui implique une sélection des embryons avant leur implantation dans l’utérus. La technique permet d’implanter uniquement les embryons ayant hérité des copies saines du gène. De ce fait, même si l’un d’eux porte la mutation MH, le DPI rend possible au couple la conception d’un enfant qui ne porte pas le gène HTT mutant, et ce que le porteur du gène soit l’homme ou la femme. Toutefois, le DPI n’est pas autorisé dans certains pays en raison des lois de protection de l’embryon, et il est aussi important de noter que les chances qu’a une grossesse d’arriver à terme après un DPI/FIV sont moins élevées que dans le cas d’une conception « naturelle ». Dans certains pays, il est possible de tester des foetus à naître, conçus naturellement, et de choisir d’avorter une fois que le statut génétique du foetus est connu.Est-il possible de s’assurer de concevoir un enfant qui ne porte pas ce
gène pathologique ?
Le diagnostic prénatal (avant la naissance) est seulement faisable lorsque les personnes le demandant remplissent certains critères médicaux et juridiques, qui dépendent de chaque pays. Il existe deux procédures standard pour le diagnostic prénatal. La première est l’amniocentèse (appelée aussi test de liquide amniotique), au cours de laquelle le liquide amniotique contenant les cellules foetales est collecté à l’aide d’une aiguille insérée à travers la paroi abdominale de la mère, en général après la 14ème semaine de grossesse. La seconde est le prélèvement des villosités choriales (tissu placentaire), pouvant être réalisé plus tôt – entre la 10ème et 13ème semaine de grossesse. Elle est cependant plus risquée pour le foetus.Puis-je faire tester mon enfant à naître ?
Il n’existe actuellement aucune thérapie pouvant traiter efficacement les causes sous-jacentes de la MH. Toutefois, la recherche fondamentale et clinique a considérablement amélioré notre connaissance de la MH ces dernières années, et plusieurs études sont maintenant en cours pour déterminer sa pathogénèse, dans le but d’identifier des médicaments qui décaleraient l’apparition de la maladie ou qui ralentiraient sa progression. Des traitements permettant d’atténuer certains symptômes de la maladie sont déjà disponibles (traitements symptomatiques), et qui améliorent la qualité de vie des patients. Ils se répartissent en traitements pharmacologiques (médicamenteux) et non pharmacologiques (non médicamenteux).Existe-t-il des traitements pour la MH ?
La chorée, la bradykinésie, l’irritabilité, l’apathie, la dépression, l’anxiété et les perturbation du sommeil sont rapportées comme étant les symptômes les plus pénibles de la MH. Il existe plusieurs options médicamenteuses pour gérer ces symptômes. Toutefois, beaucoup de médicaments peuvent engendrer des effets secondaires, et certains peuvent contrebalancer l’effet des autres. De plus, un même médicament peut avoir des effets différents sur différents individus. Le traitement doit être personnalisé par un spécialiste de la MH expérimenté, en fonction des symptômes du patient et de sa réaction aux médicaments en question.Quelles sont les options médicamenteuses pour traiter les symptômes
de la MH ?
Les traitements non pharmacologiques (tels que la psychothérapie, les thérapies cognitives, la physiothérapie, l’orthophonie, les thérapies respiratoires et l’ergothérapie) peuvent améliorer les symptômes psychologiques et physiques de la MH. Par exemple, on remarque des améliorations de l’humeur, du contrôle moteur, de l’élocution, de l’équilibre, de la déglutition, de la démarche, après de telles thérapies. Il est établi que l’exercice physique améliore à la fois la santé physique et mentale, augmentant le bien-être général, et il a été démontré que l’exercice atténue les symptômes dépressifs. De nombreuses preuves indiquent qu’il pourrait également ralentir la progression des troubles du mouvement dans la MH. Par exemple, certains programmes de physiothérapie offrent des bénéfices en termes de symptômes moteurs, démarche et équilibre. Le Groupe de Travail sur la Physiothérapie d’EHDN a publié un document de référence à l’usage des physiothérapeutes qui travaillent avec des patients MH.En quoi les traitements non pharmacologiques peuvent-ils aider ?
Beaucoup de discussions ont été menées au sujet des bénéfices d’un régime riche en vitamines, co-enzymes et autres composants, mais ils restent à être démontrés cliniquement. Toutefois, comme la perte de poids est un problème pour certains patients MH, surtout dans les stades avancés de la maladie, il est important de conserver un régime sain tout au long de la maladie. Au cours des stades plus avancés, il peut être nécessaire d’adopter un régime à hautes calories. Il peut être utile de se référer à un diététicien.Un régime alimentaire spécial peut-il atténuer les symptômes de la MH ?
Hérédité & Causes de la MH
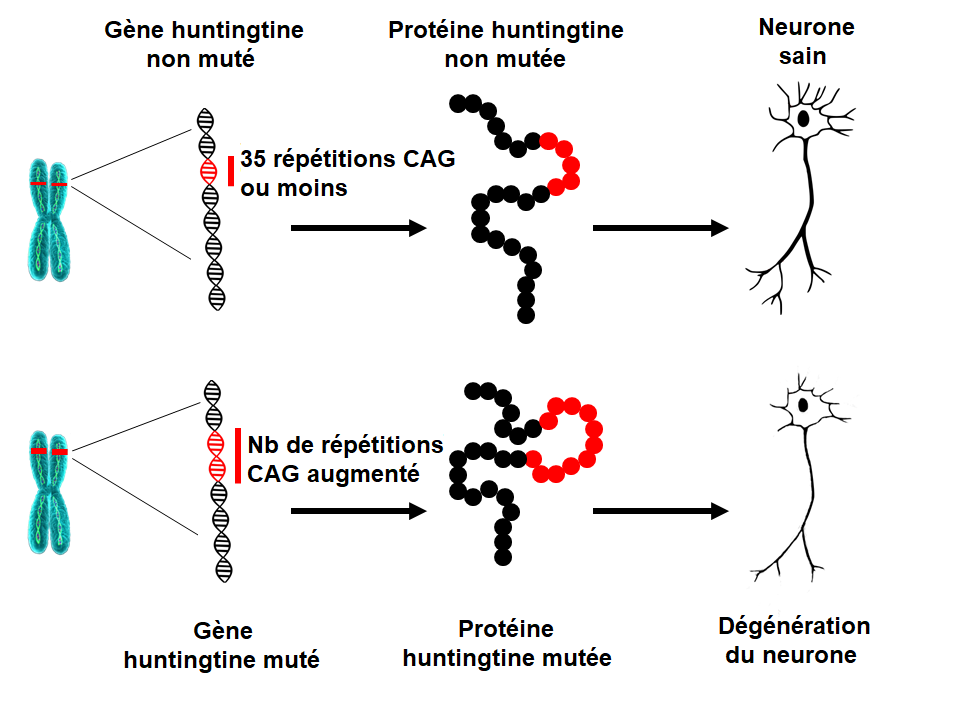
La MH est causée par un changement (une mutation) dans le gène (HTT) qui code la protéine appelée huntingtine. Le résultat de cette mutation est que le gène est traduit en une protéine de forme altérée, ayant pour conséquence le dysfonctionnement et la mort des cellules nerveuses (neurones) dans des régions spécifiques du cerveau. Les mécanismes exacts de la maladie sont multiformes et hautement complexes, de la même manière que les fonctions de la protéine huntingtine sont multiples. Les chercheurs travaillent à une meilleure compréhension des causes sous-jacentes de la maladies pour développer des thérapies pouvant modifier la maladie.Quelles sont les causes de la MH ?
En 1993, les scientifiques ont identifié la mutation qui est à l’origine de la MH. Le gène HTT est localisé sur le chromosome 4 et code pour une protéine appelée la huntingtine. Le gène contient une séquence de trois nucléotides (les unités de base de l’ADN), cytosine-adenine-guanine (CAG), qui est répétée plusieurs fois. Cette répétition de trinucléotides peut varier en longueur. Si une personne a une répétition de 40 CAG ou plus sur une copie du gène HTT, il/elle développera la MH au cours de sa vie – généralement au milieu de la vie adulte. Puisque la mutation qui cause la MH est présente dans toutes les cellules du corps depuis la conception, et peut être transmise aux générations suivantes, la MH est une maladie héréditaire.Quelle est la cause sous-jacente de la MH ?
A mesure que la répétition du nombre de CAG augmente, cette section particulière de l’ADN devient plus instable. Cela signifie que le nombre de répétitions dans cette section peut augmenter ou diminuer lorsqu’elle est transmise à la génération suivante. Du moment que la répétition du nombre de CAG sur le gène de la HTT est inférieure à 27, la section est stable. Si le nombre de répétitions est compris entre 27 et 35 (ce qu’on appelle la longueur de répétition intermédiaire), cette personne ne développera pas la MH. Cependant, une répétition de CAG de 27 ou plus est instable et susceptible d’augmenter lorsqu’elle est transmise à la génération suivante, ce qui signifie que ces enfants présentent un risque de développer la MH. Les personnes ayant un nombre de CAG entre 36 et 39 peuvent développer la MH, mais seulement très tard dans la vie, ou ne pas la développer. Il s’agit de l’intervalle de répétitions à pénétrance réduite. Lorsque le nombre de répétitions de CAG est supérieur à 39, la personne développera la MH au cours de sa vie – le plus souvent au milieu de la vie adulte. Dans de rares cas, l’expansion CAG peut être exceptionnellement longue, conduisant à une apparition de la maladie au cours de l’adolescence ou de l’enfance (MH juvénile). Les patients qui développent la maladie avant l’âge de 10 ans possèdent souvent une répétition de plus de 80 CAG.Que signifie la longueur de la répétition CAG sur le gène de la HTT ?
Longueur de la répétition CAG
Cause de la maladie?
Conséquences
pour la
descendance?
Nom
En-dessous de 27
Non
Aucune
Répétition de longueur normale
27-35
Non
Les répétitions de 27 et plus peuvent être instables et peuvent augmenter lorsque transmises à la descendance
Répétition de longueur intermédiaire
36-39
Peut-être
Oui, la descendance a une probabilité de 50% d'hériter du gène muté
Répétition d'une longueur de pénétrance réduite
40 et plus
Oui
Oui, la descendance a une probabilité de 50% d'hériter du gène muté
Répétition d'une longueur de pénétrance totale
Les gènes se trouvent dans nos chromosomes à l’intérieur de chaque cellule de notre corps. Un gène est une portion d’ADN qui contient le code pour une protéine particulière ; l’ADN est transcrit en ARN messager (ARNm), qui est ensuite traduit en protéine. La plupart du temps, tout le monde hérite de deux copies de chaque gène – l’une provenant de la mère, l’autre du père. Dans la MH, le gène important est celui de la HTT, codant pour la protéine huntingtine. Lorsqu’un enfant hérite d’une version mutée du gène HTT, il développera alors également la MH. Le parent lui-même peut déjà avoir la maladie ou celle-ci pourra se développer à un âge ultérieur.Qu'est-ce qu'un gène ?
correspondant à ce gène. Par conséquent, les gènes fonctionnement comme des schémas directeurs – des ensembles d’instructions qui dictent aux cellules comment construire des protéines spécifiques. Le gène HTT contient des instructions pour construire la protéine huntingtine. Les protéines sont les molécules qui réalisent le travail à l’intérieur des cellules – elles réalisent un grand nombre de processus, tels que les réactions enzymatiques ou le support structurel. Si une protéine dysfonctionne ou est manquante en raison d’une mutation dans le gène qui la code, cela peut affecter la cellule et à terme l’ensemble de l’organisme, causant parfois des maladies.Qu'est-ce qu'une protéine ?
La huntingtine est une protéine très large qui est produite, ou « exprimée » à des degrés variés dans chaque cellule du corps humain ; les plus hauts niveaux se retrouvent dans le cerveau. La huntingtine semble être une protéine très importante car son absence est léthale chez les embryons de souris. Au bout de l’une des extrémités de la protéine huntingtine se trouve une portion de répétitions d’un acide aminé particulier qui s’appelle la glutamine. Cette caractéristique distinctive, connue comme étant une répétition de polyglutamine, consiste normalement en une répétition allant jusqu’à 35 unités de glutamine. Chez les personnes porteuses de la mutation MH, elle contient cependant au moins 36 répétitions, et c’est cette expansion de polyglutamine qui conduit au dysfonctionnement de la protéine.La protéine huntingtine
La MH est une maladie héréditaire dominante. Cela signifie qu’une personne née avec une copie du gène HTT mutante développera la MH même s’il/elle porte également une copie normale du gène. Un porteur de la mutation MH, qu’il soit symptomatique ou non, peut transmettre une copie soit normale soit mutante du gène avec une probabilité de 50% pour chacune (à condition qu’il/elle ne porte la mutation que sur l’une des deux copies du gène HTT). Des techniques médicales permettent de s’assurer qu’une personne affectée ne transmette à son enfant que le gène HTT normal. D’autre part, une personne n’ayant pas hérité du gène HTT ne développera pas la maladie, et son enfant ne présentera pas non plus de risque de la développer. La mutation MH ne peut pas sauter de génération. Toutefois, il peut arriver qu’un porteur de la mutation décède avant d’avoir manifesté des symptômes, et que ses enfants ne sachent pas qu’ils présentent un risque de développer la maladie.Comment la MH est-elle transmise ?
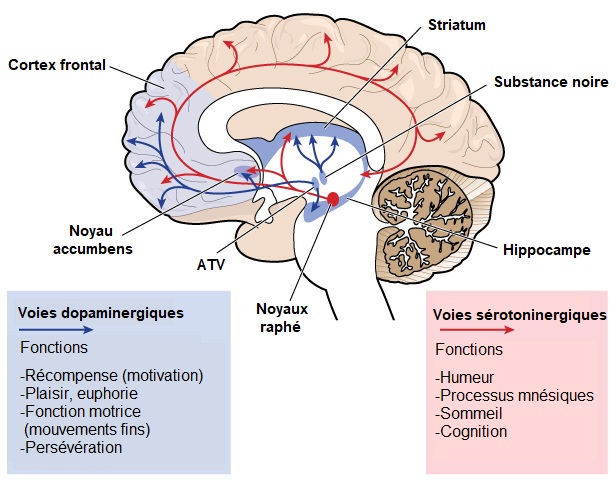
Certaines fonctions cérébrales comme la capacité à se mouvoir, réfléchir et parler se détériorent graduellement dans la MH à mesure que les cellules nerveuses cruciales se dégradent et meurent. La partie du cerveau la plus affectée par la MH est le striatum, qui est un composant des ganglions de la base et se situe profondément dans la région centrale du cerveau. Le striatum est principalement impliqué dans la planification et le contrôle des mouvements, mais aussi dans beaucoup d’autres processus, incluant la cognition et les émotions. A mesure que la MH progresse, cela affecte le cortex (la partie extérieure et plissée du cerveau), contribuant à la détérioration cognitive. En général, la MH provoque avec le temps une atrophie de l’ensemble du cerveau, altérant les capacités fonctionelles générales de l’individu.Les régions du cerveau concernées par la MH
La MH dans la vie quotidienne
Être testé positivement pour la mutation MH peut affecter différents aspects de la vie d’une personne, comme la décision d’avoir ou non des enfants, la planification de l’avenir, repenser les priorités, l’achat d’un logement adéquat, et informer les autres membres de la famille qu’ils pourraient également être à risque pour la MH. Les jeunes adultes en particulier peuvent avoir besoin de considérer les implications d’un test positif par rapport à leurs études, leur éducation et leur emloi. A mesure que la maladie progresse, elle affecte graduellement la capacité d’une personne à vivre de manière indépendante. Le travail, la vie sociale et les activités de la vie quotidienne deviennent problématiques et les patients peuvent devenir de plus en plus dépendants de l’aide de leurs proches et des professionnels de santé et des travailleurs sociaux. Les groupes de défense des patients locaux et les centres cliniques peuvent être contactés à tout moment, et apporteront leur soutien.Comment la MH affecte-t-elle la vie quotidienne ?
Les stratégies efficaces pour faire face à la MH doivent être personnalisées et dépendent de la personne affectée, le stade de la maladie et le contexte familial. La MH se développe très lentement, de manière à ce que, en général, la personne a le temps de s’adapter aux changements qu’elle amène. Pour les aidants et les proches, une meilleure compréhension des détériorations cognitives et comportementales associées à la maladie peuvent les aider à développer des stratégies pour s’accommoder de ces changements et maintenir une bonne relation avec la personne affectée. Des informations et des conseils utiles peuvent être donnés par les spécialistes de la MH et les groupes de défense des patients.Existe-t-il des stratégies pour mieux faire face à la MH ?
En allant sur cette page, vous trouverez une liste de coordinateurs linguistiques qui peuvent vous aider. Vous pouvez également utiliser le formulaire de contact que vous trouverez ici.Comment contacter EHDN ?
Vous pouvez prendre un rendez-vous en étant orienté par votre médecin traitant ou en contactant votre coordinateur linguistique local d’EHDN qui vous aidera.Comment obtenir un rendez-vous avec un spécialiste ?
Vous pouvez obtenir un avis indépendant sur la MH à partir des groupes de défense des patients dans votre pays.Est-il possible de parler à un spécialiste sans aller à l'hôpital ?
EHDN joue un rôle clé dans l’étude clinique mondiale Enroll-HD. Il s’agit d’une étude observationnelle qui n’implique pas d’intervention. Cela signifie qu’elle ne teste pas de thérapie expérimentale à proprement dit. Les participants se soumettent à des évaluations cliniques au cours de leur visite annuelle à l’hôpital et ils peuvent être éligibles pour participer à des essais cliniques sur des traitements symptomatiques ou modifiant le cours de la maladie, si de tels traitements deviennent disponibles. Enroll-HD est accessible dans beaucoup de centres d’études MH dans le monde entier. Pour savoir s’il y a un centre près de chez vous, vous pouvez consulter cette page ou vous rapprocher de votre coordinateur linguistique EHDN, qui pourra vous renseigner au sujet des activités de recherche dans votre région. Les groupes de défenses des patients au niveau local pourront vous fournir des informations générales sur la participation à la recherche. Pour plus d’informations au sujet de la recherche sur la MH, vous pouvez consulter cette page ou visiter le site internet HDBuzz qui contient des informations écrites en langage vulgarisé par des chercheurs spécialistes de la MH et traduites dans la plupart des langues.Comment puis-je m'impliquer dans la recherche sur la MH ?
Oui, un certain nombre de groupes de défense des patients offrent un soutien aux personnes et aux familles affectées par la MH. Vous pouvez les contacter en passant par votre médecin traitant ou spécialiste MH, ou vous pouvez les contacter directement. La European Huntington’s disease Association (EHA) tient une liste de groupes de défense des patients qui pourrait vous être utile.Existe-t-il des groupes de soutien spécialisés sur la MH ?
Pour toute autre question, veuillez contacter votre coordinateur linguistique d’EHDN ou votre groupe de défense des patients local